 Any person’s dream is to stay young as long as possible. We don’t want to get old and sick, we are afraid of everything - cancer, Alzheimer's disease, heart attack, stroke ... It's time to figure out where the cancer comes from, is there a connection between heart failure and Alzheimer's disease, infertility and hearing loss. Why do antioxidant supplements sometimes do more harm than good? And most importantly: can we live long and without disease, and if so, how?
Any person’s dream is to stay young as long as possible. We don’t want to get old and sick, we are afraid of everything - cancer, Alzheimer's disease, heart attack, stroke ... It's time to figure out where the cancer comes from, is there a connection between heart failure and Alzheimer's disease, infertility and hearing loss. Why do antioxidant supplements sometimes do more harm than good? And most importantly: can we live long and without disease, and if so, how?
We have tiny “energy stations” in our bodies - mitochondria. They are responsible for our health and well-being. When they work well, we are not lacking in energy. And when it’s bad, we suffer from diseases. Dr. Lee Noe reveals a secret: diseases that seem unrelated at first glance: diabetes, cancer, schizophrenia, chronic fatigue, Parkinson's disease and others - have a common nature.
Today we know how to improve the functioning of mitochondria, which provide the body with energy by 90%. In this book you will find relevant information about nutrition, lifestyle, ketogenic diet and supplements that restore health to mitochondria, and therefore to us.
Excerpt. Mitochondrial Syndrome
I am embarrassed to admit it, but I was a spectator of the reality show "The Bachelor". I was very impressed with the third episode of season 17 (January 2013), in which Sin (a bachelor) and Ashley (an applicant) went to meet two girls suffering from mitochondrial disease. For many of you, if you watched the episode, this was the first acquaintance with mitochondrial syndrome (mitochondrial syndrome is a complex of diseases associated with congenital damage to the mitochondria). However, this group of diseases is being studied more and more qualitatively as the technology of genetic testing and genetic sequencing becomes easier, cheaper and more affordable.
Until the early 80s of the last century, when the human mitochondrial genome was completely sequenced, reports of mitochondrial diseases were rare. The situation has changed with the possibility of decoding mtDNA of many patients. This has led to a sharp increase in the number of registered patients suffering from hereditary mitochondrial diseases. These include approximately one in five (or even two and a half) thousand people. Here we do not include individuals with unexpressed forms of mitochondrial disease. In addition, the list of signs of mitochondrial syndrome has grown sharply, which indicates the chaotic nature of these diseases.
Mitochondrial diseases are characterized by extremely complex genetic and clinical pictures, which are a mix of a very wide range of existing diagnostic categories. Inheritance patterns here sometimes obey, and sometimes disobey, Mendel’s laws. Mendel described patterns of inheritance of traits through normal nuclear DNA genes. The probability of the appearance of a genetic trait or hereditary disease is easily calculated based on a quantitative forecast of the results of the splitting of offspring according to different qualitative characteristics by randomly inheriting one of two copies of the same gene from each of the parents (as a result, each of the offspring receives two copies of each gene). In cases where the mitochondrial syndrome is caused by a defect in nuclear genes, the corresponding inheritance patterns do follow the rules of Mendel. However, there are two types of genomes that provide mitochondrial function: mitochondrial DNA (transmitted only through the maternal line) and nuclear DNA (inherited from both parents). As a result, inheritance types range from autosomal dominant to autosomal recessive, as well as to maternal transmission of genetic material.
The situation is further complicated by the fact that complex interactions are built in the cell between mtDNA and nDNA. As a result, the same mtDNA mutations can cause dramatically different symptoms from siblings living in the same family (they can have different nuclear DNAs with identical mtDNAs), while mutations can cause identical symptoms. Even twins with the same diagnosis may have radically different clinical pictures of the disease (specific symptoms depend on which tissues are affected by the pathogenic process), while people with mutations may suffer from similar symptoms lining up in the same picture of the disease.
Be that as it may, a large number of mtDNA variations exist in the maternal egg, and this fact devalues all predictions regarding the results of genetic inheritance. The nature of this group of diseases is so chaotic that the set of symptoms corresponding to these diseases can vary from decade to decade and even differ for siblings with identical mitochondrial DNA mutations. Moreover, sometimes the mitochondrial syndrome can simply disappear, despite the fact that it was (or should have been) inherited. But such happy cases are rare, and most often mitochondrial diseases progress. In the table. 2.2 and 2.3 present diseases and symptoms associated with mitochondrial dysfunction, as well as the genetic factors of these diseases. Currently, science knows over 200 types of mitochondrial mutations. The research results suggest that many degenerative diseases are caused by mutations of this kind (this means that we must reclassify a huge number of diseases, translating them into the category of mitochondrial diseases).
As we know, these mutations can lead to the fact that mitochondria cease to perform the function of energy production, as a result of which cells can interrupt their work or die. All cells (except for red blood cells) contain mitochondria, and, accordingly, mitochondrial syndrome affects multicomponent and very different systems of the body (simultaneously or sequentially).
Table 2.2. Signs, symptoms, and diseases caused by mitochondrial dysfunction
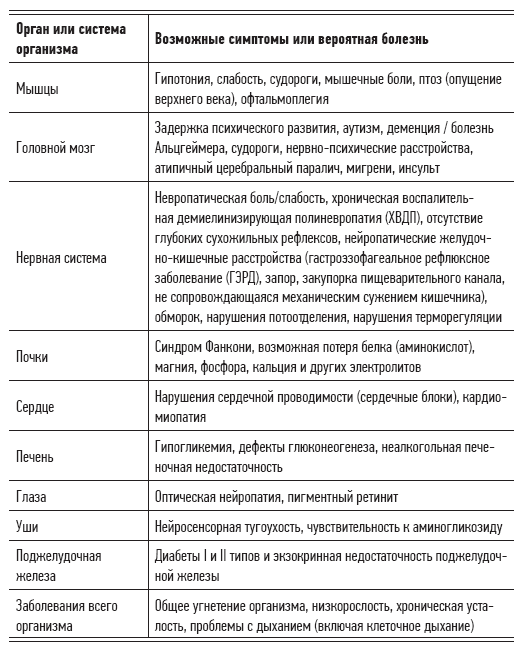
Table 2.3. Congenital diseases caused by mitochondrial dysfunction
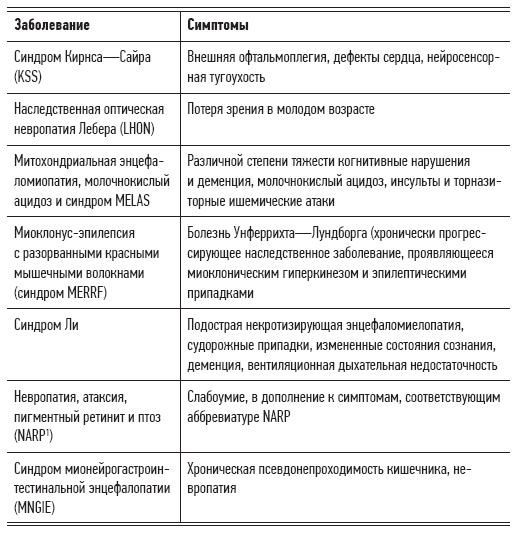
Of course, some organs or tissues need more energy than others. When the energy needs of a particular organ cannot be fully satisfied, the symptoms of mitochondrial syndrome begin to appear. First of all, they affect the functions of the brain, nervous system, muscles, heart, kidneys and endocrine system, that is, all organs that require a large amount of energy for normal operation.
Acquired mitochondrial dysfunction
As our understanding of mitochondrial function and dysfunction grows, we begin to create a long list of diseases based on mitochondrial dysfunction and to clarify the mechanisms of the occurrence and development of these ailments. Some recent studies suggest that every 2500th person suffers from mitochondrial syndrome. However, if you carefully study the list below, you will agree that with a high degree of probability mitochondrial diseases (congenital or acquired) will soon be detected in every twenty-fifth or even every tenth inhabitant of the Western countries.
- Type II diabetes
- Cancer
- Alzheimer's disease
- Parkinson's disease
- Bipolar Affective Disorder
- Schizophrenia
- Aging and decrepitude
- Anxiety disorder
- Non alcoholic steatohepatitis
- Cardiovascular diseases
- Sarcopenia (loss of muscle mass and strength)
- Exercise intolerance
- Fatigue, including chronic fatigue syndrome, fibromyalgia, and myofascial pain
At the genetic level, very complex processes are associated with all this. The energy strength of a particular person can be determined by examining the innate abnormalities of his mitochondrial DNA. But this is only the starting point. Over time, acquired defects of mtDNA accumulate in the body, and after a particular organ crosses a certain threshold, it begins to junk or becomes susceptible to degeneration (each organ has its own threshold of patience, which we will discuss in more detail).
Another complication is that each mitochondria includes up to ten copies of mtDNA, and each cell, each tissue and each organ has many mitochondria. It follows that our body does not count defects in copies of mtDNA. Dysfunction of a particular organ begins when the percentage of defective mitochondria living in it exceeds a certain value. This phenomenon is called the threshold effect36. Each organ and each tissue is subject to specific mutations and is characterized by its own mutational threshold, energy requirements and resistance to exposure from free radicals. The combination of these factors determines what the reaction of the living system to genetic disorders will be.
If only 10% of mitochondria are defective, 90% of the remaining normal generators of cellular energy can compensate for the dysfunction of their “colleagues”. Or, for example, if the mutation is not very serious, but has affected a large number of mitochondria, the cell can still function normally.
There is also the concept of segregation of defective mitochondria: during cell division, its mitochondria are randomly distributed between two daughter cells. One of these cells can receive all mutated mitochondria, while the other can acquire all full-fledged “power plants” (of course, intermediate variants are more likely). Cells with dysfunctional mitochondria will die during apoptosis, and healthy ones will continue to do their job (one of the explanations for the sudden and unexpected disappearance of mitochondrial syndrome). The phenomenon of differences in the DNA sequence of mitochondria (or plastids) in the same organism, often even in one cell, when some mitochondria, for example, may contain some pathological mutation, while others do not, is called heteroplasmy. The degree of heteroplasmy differs even among members of the same family. Moreover, the level of heteroplasmy can vary even within the same organism depending on a specific organ or specific cell, which leads to a very wide range of manifestations and symptoms of a particular mitochondrial disease.
In the body of a growing embryo, as cells divide, mitochondria with mutations fill organs and tissues that differ from each other in terms of their energy needs. And if mutated mitochondria populate cells in large numbers, which eventually turn into metabolically active structures (for example, the brain or heart), then the corresponding organism further has problems with the quality of life (if at all viable). On the other hand, if a mass of dysfunctional mitochondria accumulates primarily in cells with a low metabolic rate (say, skin cells that regularly replace one another), then the carrier of such mitochondria may never know about their genetic predisposition to mitochondrial syndrome. In the above episode from The Bachelor, one of the girls with mitochondrial disease seemed quite normal, while the other was obviously suffering from a serious ailment.
Some mitochondrial mutations spontaneously develop with age as a result of the formation of free radicals during normal metabolism. What happens next depends on a number of factors. For example, if a cell filled with dysfunctional mitochondria divides at high speed, as stem cells doing tissue regeneration do, defective energy generators will actively expand. If the weakened cell no longer divides (suppose we are talking about a neuron), then the mutations will remain within this cell only, which, however, does not exclude the possibility of a successful random mutation. So, it is precisely the complexity of the genetic basis of the mitochondrial syndrome that explains the fact that the depletion of bioenergy resources of the body caused by mutations of the mitochondria manifests itself in a wide range of diverse and complex diseases and symptoms.
We must also remember that there are many genes outside of mtDNA that are responsible for the normal functioning of mitochondria. If the mutation affects genes encoding RNA, then the consequences are usually very serious. In those cases when a child receives a mutated transcription factor of mitochondria from any of the parents during their conception (recall that transcription factors are proteins that control the process of mRNA synthesis on the DNA matrix by binding to specific DNA sites), then all mitochondria will undergo pathogenic effects organism. However, if the mutation refers only to specific transcription factors that are activated only in certain organs or tissues or in response to the release of a specific hormone, then the corresponding pathogenic effect will be exclusively local.
A wide range of mitochondrial diseases and their manifestations is a serious problem for physicians (both theoretical and practical), including the actual impossibility to predict the development of mitochondrial syndrome. There are so many mitochondrial diseases that it is difficult for them all to simply give names, and yet many of them have not yet been discovered. Even a number of known degenerative diseases (diseases of the cardiovascular system, oncological diseases, various forms of dementia, etc.) are referred to by modern science as mitochondrial dysfunction.
It is important to realize that although there is no full-fledged treatment for mitochondrial diseases, many people with these ailments (especially when it comes to mild or moderate forms of the disease) can live a long and full life. However, for this it is necessary to work systematically, using the knowledge that has appeared at our disposal.
about the author
Lee Nou is a licensed naturopathic practitioner from Canada and has received several awards. Colleagues know him as a visionary entrepreneur, strategist and doctor. Lee served as a medical consultant, scientific expert and director of research and development in large organizations. In addition to research at his company, he is also a consultant in the field of natural health products and nutritional supplements, and is also a member of the editorial and advisory board of Alive Magazine, Canada's most widely read health magazine. He calls home the Greater Toronto area, where he lives with his wife and their two sons, and is particularly interested in strengthening natural health and protecting the environment.
»More details on the book can be found on the publisher’s website
» Contents
» Excerpt
25% discount on coupon for Khabrozhitel - Mitochondria
Upon payment of the paper version of the book, an electronic book is sent by e-mail.