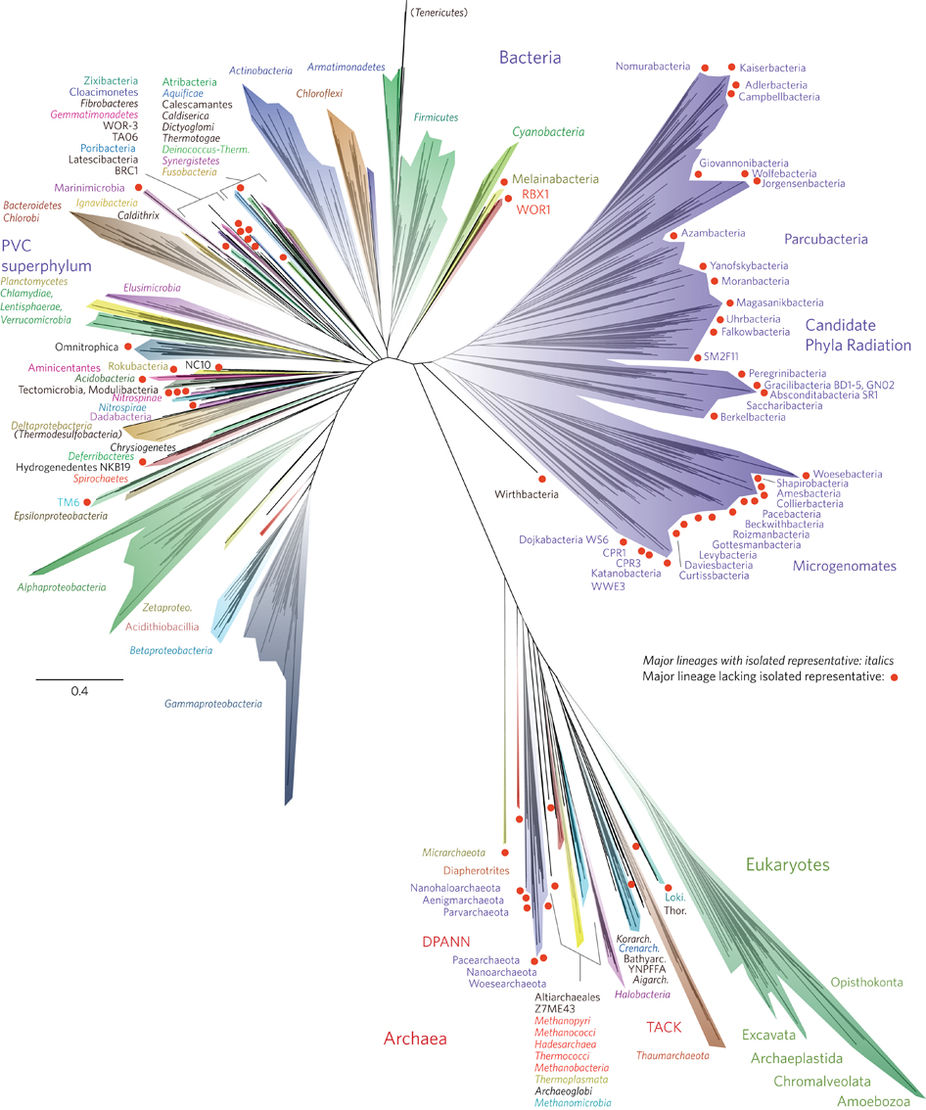
KDPVには何がありますか?
現在存在する最も完全な生命の木 。 すべての多細胞生物は、右下隅の緑色のスポットに収まり、そのわずかな部分を占めます。また、すべての動物とキノコ(オピストコンタと総称)は、このスポットの右端の棚にあります。 これはほぼ地球上の生命の多様性であり、それはそれを研究する過程で生成されるデータの量です。
ところで、赤い丸は、誰も見たことがない単細胞生物のグループを強調しています。 ペトリ皿や独創的なバイオリアクターで培養が成長する研究室はありません。 それらの存在は、すべてのDNAが培地から抽出され、一度に分析されるメタゲノム研究から近年になって初めて知られるようになりました。 しかし、これはそうです。 この写真は、ここ数年で最も驚くべきツリーとしてのみ記事に含まれており、記事を理解するために重要ではありません。
ところで、赤い丸は、誰も見たことがない単細胞生物のグループを強調しています。 ペトリ皿や独創的なバイオリアクターで培養が成長する研究室はありません。 それらの存在は、すべてのDNAが培地から抽出され、一度に分析されるメタゲノム研究から近年になって初めて知られるようになりました。 しかし、これはそうです。 この写真は、ここ数年で最も驚くべきツリーとしてのみ記事に含まれており、記事を理解するために重要ではありません。
どうする
まず、何が起こっているのか、何を達成しようとしているのかを明確に述べます。 私たちは、この遺伝子または人がどこから来たのか、そしてどのように進化したのかを知りたいのです。 またはジャガイモ、または単細胞藻類、どの生物の方法もほぼ同じです。 生物学的データベースでは、他の生物の関連配列を見つけ、それらを特定の 魔法の プログラムに入れて、系統樹を出力として取得できます。 問題は、関連する数千のシーケンスがある可能性が高いことです。 そして、 最尤法またはベイジアン系統学による系統樹の構築はNP完全なタスクであり、解決策は、大まかに言って、進化モデルのさまざまな木とパラメーターをソートすることです。 通常、数百の中程度の遺伝子に対して64 GbのRAMを搭載した2つの16コアAMD Opteron 6276プロセッサーでは、数日から1週間かかります。 系統解析の多項式法がありますが、それらはそれほど正確ではなく、それらに基づいた記事をまともなジャーナルに公開するのは非現実的です。
したがって、このセットから正しい選択を行う必要があります。指定されたサイズの比較的小さなシーケンスのセットを選択し、そこから母集団全体について結論を導き出します。 次に、いくつかの系統学的用語が使用されますが、説明はネタバレです。
主な概念と用語
まず、系統樹とは何ですか? これは、単一の祖先から連続した分岐を経て多くの現代のDNAまたはタンパク質配列の出現を記述するバイナリツリーです。 そのような祖先によって結合された遺伝子(またはタンパク質)は、互いに相同です。 何十億年も前に存在したとしても、単一の祖先があったに違いありません。そうでなければ、分析全体が意味をなしません。
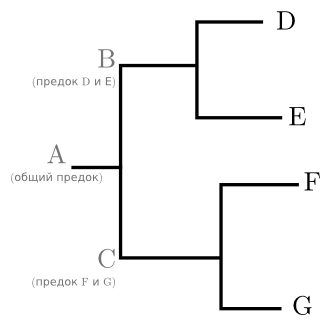
例(下図):子孫BとCを持つ祖先遺伝子Aがあった。Bは子孫DとE、C-FとGを残した。4つのシーケンスD、E、FおよびGのみが観察される。 AからCまでの先祖は昔々存在し、今では姿を消しています。 したがって、それらについては、単に現代のシーケンスに基づいて、主に「そのようなもの、そのようなもの、そのようなものの共通の祖先」などの用語で話すことができます。 共通の祖先によって結合されたツリーの一部は、 トレジャーと呼ばれます。 この用語は、グラフの実際の部分と、グラフの両端にあるシーケンスの両方に適用できます。 たとえば、図のツリーには、宝物(D、E)と(F、G)があります。 ツリーの名目上の宝物は内部ノードと同じくらいですが、通常、研究者は自分の仕事に重要ないくつかの宝物にしか興味がありません。
誰もが種の進化について考えるのに慣れているのに、なぜシーケンスの進化について話しているのでしょうか。 最終的に、DNAの小片ではなく、生物全体が餌を与え、増殖し、死滅します。 これは一般的に正しいです。 「生命の木」はもともと種のために発明されたもので、系統樹はその進化を完全に記述しています。 しかし、シーケンスツリーを具体的に説明する理由は2つあります。まず、バイオインフォマティクスの分野としての系統発生学は、ほとんどの場合、シーケンスを扱います。 特性の組み合わせによる生物の進化は、複雑で形式化するのが難しいプロセスであり、さらに、数百万年前に未知の要因の影響下で行われ、一般に誰とかはあまり明確ではありません。 たとえば、最初の真核生物が正確に何であったかについての議論は、タイムマシンの発明以前に終わらない可能性があります。 一方、文字列内の文字の置換、挿入、削除は、数学モデリングに非常に適しています。
第二に、個々の遺伝子の進化は、それが存在する種の進化とは異なる場合があります。 最も単純なシナリオは上のツリーで説明されています-研究された種の共通の祖先は研究された遺伝子のコピーを1つ持ち、進化の間にそれは単純に継承され、小さな突然変異を徐々に蓄積しました。 そのような遺伝子は、 オルソログによって互いに帰属しています。 定義により、オーソログは異なる種の遺伝子であり、共通の祖先の単一の遺伝子に戻ります。
しかし、祖先Cが研究対象の遺伝子の複製を持っていたと仮定します。 1つの遺伝子があり、互いに独立して遺伝し、変異し始めた2つの同一の遺伝子がありました。 遺伝子のコピーの1つが以前と同じように機能し続け、2つ目(古い機能を実行せずに古い機能を実行する人がいるため)が変異し、新しい利点がもたらされる可能性があります。 しかし、種Eまたはその先祖の1つは、この遺伝子を完全に失いました。 これは、遺伝子がそれほど重要でない場合に起こります。 種間の関係は変更されていませんが、ツリーは次の形式を取ります。
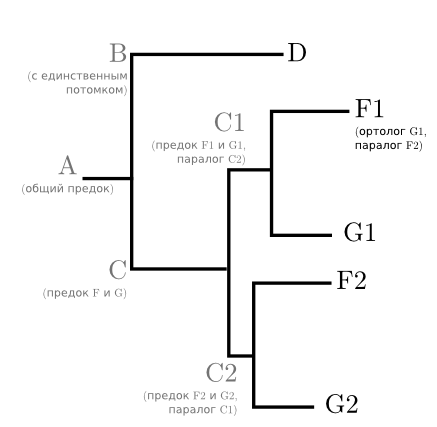
このツリーに基づいて、種の進化について話すことはすでに非常に困難です。 Eはまったく存在せず、FとGはそれぞれ2回検出されます。 しかし、それにより、* 1および* 2遺伝子がいつ出現し、どこまで行ったかを理解することができます。 遺伝子F1とG1は互いにペアになっています。同じ種の遺伝子で、祖先の唯一の遺伝子に遡ります。
複製と削除に加えて、他のイベントも発生します:遺伝子は親からではなく、他の非関連生物(水平遺伝子伝達またはHGTと呼ばれます)から取得されたり、他の遺伝子の一部が1つの遺伝子に追加されたり、逆の場合もあります、断片が切り取られ、時にはいくつかの遺伝子が1つに結合されます。 一般に多くの興味深いことが起こっています。 そのため、すべての相同配列が分析に投入された場合にどのような結果が得られるかは事前にはわかりません。
例(下図):子孫BとCを持つ祖先遺伝子Aがあった。Bは子孫DとE、C-FとGを残した。4つのシーケンスD、E、FおよびGのみが観察される。 AからCまでの先祖は昔々存在し、今では姿を消しています。 したがって、それらについては、単に現代のシーケンスに基づいて、主に「そのようなもの、そのようなもの、そのようなものの共通の祖先」などの用語で話すことができます。 共通の祖先によって結合されたツリーの一部は、 トレジャーと呼ばれます。 この用語は、グラフの実際の部分と、グラフの両端にあるシーケンスの両方に適用できます。 たとえば、図のツリーには、宝物(D、E)と(F、G)があります。 ツリーの名目上の宝物は内部ノードと同じくらいですが、通常、研究者は自分の仕事に重要ないくつかの宝物にしか興味がありません。
誰もが種の進化について考えるのに慣れているのに、なぜシーケンスの進化について話しているのでしょうか。 最終的に、DNAの小片ではなく、生物全体が餌を与え、増殖し、死滅します。 これは一般的に正しいです。 「生命の木」はもともと種のために発明されたもので、系統樹はその進化を完全に記述しています。 しかし、シーケンスツリーを具体的に説明する理由は2つあります。まず、バイオインフォマティクスの分野としての系統発生学は、ほとんどの場合、シーケンスを扱います。 特性の組み合わせによる生物の進化は、複雑で形式化するのが難しいプロセスであり、さらに、数百万年前に未知の要因の影響下で行われ、一般に誰とかはあまり明確ではありません。 たとえば、最初の真核生物が正確に何であったかについての議論は、タイムマシンの発明以前に終わらない可能性があります。 一方、文字列内の文字の置換、挿入、削除は、数学モデリングに非常に適しています。
第二に、個々の遺伝子の進化は、それが存在する種の進化とは異なる場合があります。 最も単純なシナリオは上のツリーで説明されています-研究された種の共通の祖先は研究された遺伝子のコピーを1つ持ち、進化の間にそれは単純に継承され、小さな突然変異を徐々に蓄積しました。 そのような遺伝子は、 オルソログによって互いに帰属しています。 定義により、オーソログは異なる種の遺伝子であり、共通の祖先の単一の遺伝子に戻ります。
しかし、祖先Cが研究対象の遺伝子の複製を持っていたと仮定します。 1つの遺伝子があり、互いに独立して遺伝し、変異し始めた2つの同一の遺伝子がありました。 遺伝子のコピーの1つが以前と同じように機能し続け、2つ目(古い機能を実行せずに古い機能を実行する人がいるため)が変異し、新しい利点がもたらされる可能性があります。 しかし、種Eまたはその先祖の1つは、この遺伝子を完全に失いました。 これは、遺伝子がそれほど重要でない場合に起こります。 種間の関係は変更されていませんが、ツリーは次の形式を取ります。
このツリーに基づいて、種の進化について話すことはすでに非常に困難です。 Eはまったく存在せず、FとGはそれぞれ2回検出されます。 しかし、それにより、* 1および* 2遺伝子がいつ出現し、どこまで行ったかを理解することができます。 遺伝子F1とG1は互いにペアになっています。同じ種の遺伝子で、祖先の唯一の遺伝子に遡ります。
複製と削除に加えて、他のイベントも発生します:遺伝子は親からではなく、他の非関連生物(水平遺伝子伝達またはHGTと呼ばれます)から取得されたり、他の遺伝子の一部が1つの遺伝子に追加されたり、逆の場合もあります、断片が切り取られ、時にはいくつかの遺伝子が1つに結合されます。 一般に多くの興味深いことが起こっています。 そのため、すべての相同配列が分析に投入された場合にどのような結果が得られるかは事前にはわかりません。
「正しい選択」を次のように定義します。セットのすべてのシーケンスを含むツリーに、関心のある一定数の宝物があるようにします。 生物学的な意味は何であれ、 主なものは、それらが基本的に単系統であり(つまり、それぞれ単一の祖先に戻る)、基本的にツリーの残りの部分から多かれ少なかれ長い枝によって分離されていることです。 各宝物は、そのシーケンスのサブセットに対応しています。 これらの各サブセットが少なくとも1つのシーケンスで表されるように、正しいサンプルを検討します。
しないこと
最も明白なアイデアは、シーケンスをランダムに取得し、選択が多かれ少なかれ正しいことを期待することです。 しかし、チャンス-それはチャンスです。 多分それは正しいでしょうか、そうでないかもしれません。 生物圏は不均一に研究されているため、実際にはそうではないでしょう。 男性と多くのモデルオブジェクト(ショウジョウバエハエ、パン酵母、大腸菌、大腸菌など)が詳細に研究されており、可能なすべてが繰り返しシーケンスされています。 経済的または医学的に重要なオブジェクト(たとえば、米やHIV)および重要なオブジェクトの最も近い親類(たとえば、人の親類としての霊長類)は、少し悪いだけでなく、非常によく研究されています。 しかし、単細胞プランクトンは、それがどれほど多様であっても、誰にとってもほとんど関心がなく、比較的まれにしか配列されません。 したがって、100の相同遺伝子のセット、動物の30の配列、植物の30の配列、菌類の20の配列、および他のすべての真核生物グループの1つまたは2つの配列がよく現れます。
ランダムなサンプルの場合、グループ全体の比率が維持されます。 しかし、保存する必要はありません。生物学的現実ではなく、特定の分類群に関係する科学者の数と資金を反映しています。 小さなグループを含め、できるだけ多くのグループにリーチする必要があります。
さて、次のアイデア-分類学的メタ情報(どの生物がどのシーケンスを持っているか)がすでにあるので、それを使用してみませんか? 1つの人間の遺伝子を取り、ほぼ均一に他の動物のダースの遺伝子、真菌のダースの遺伝子などを取ります。 アイデアは悪くありません、 人々はそれをしますが、普遍的ではありません。 元のセットのすべての遺伝子が互いにオルソログである場合、つまり、それらの進化が所有者の進化と完全に一致する場合、そうすることが最善です。 しかし、これは常にそうではありません。 たとえば、写真を見てください:
真核生物のキチンシンターゼツリー 
これは、 昨年の記事からのキチンシンターゼ(キチン合成の原因となるタンパク質)の進化です。 ご覧のとおり、マッシュルームタンパク質(真菌クラス*)は単一の宝を形成しません。 代わりに、ツリーの2つの異なる端に散在しています。 同じことが他の生物でも起こります。たとえば、ほとんどの卵菌種は、4つの卵菌の宝物のそれぞれから1つの遺伝子を持っています。 単純化のために宝物が2つしかないことを受け入れたとしても、それらはまだ考慮に入れられなければなりません。
条件付きの「10個のキノコ遺伝子」は、クレードの1つのダース遺伝子ではなく、1つのクレードのいくつかの遺伝子と別のクレードのいくつかの遺伝子である必要があります。 同じことが残りのグループにも当てはまります。
キチンシンターゼはかなり複雑な進化をしており、ほとんどの遺伝子では木がはるかに単純になります。 しかし、ツリーの複雑さは、構築される前に推定することはできません。したがって、ユニバーサルサンプリング手法は、そのようなデータを処理できるはずです。
最後に、3番目のアイデア。 初めのどこかで、系統解析の迅速で信頼性の低い方法がさりげなく言及されました。 彼らの助けを借りて何らかの種類の木を構築し、その中の宝物を選択せず、すでにそれらのサンプルを採取しないのはなぜですか? はい、ツリーはあまり正確ではありませんが、ほとんどの場合、実際のツリーとほぼ同じです。 このようなツリーに基づいて選択を行い、すでに適切に計算できます。 この形では、「宝物の強調表示」は重要な作業であるため、このアイデアはあまり実用的ではありません。 宝物は実際にはツリー内のノードと同じ数であり、どのノードがユーザーの興味を引くかは事前にはわかりません。 しかし、この考えに基づいて構築できます。
私たちは何をしますか
ツリーをすばやく取得する最も一般的な方法の1つは、シーケンスの距離行列を構築し(生物学的シーケンスの距離メトリックは非常に異なる)、それを使用して、何らかのNJまたはUPGMAを使用してツリーを構築することです。 もちろん、距離行列の構築は、シーケンス数から時間とメモリが2次ですが、MLやベイズの系統発生学とは異なり、少なくとも多項式です。
距離行列に基づいてツリーを構築しなくても、できるだけ多くのシーケンスを含むサンプルを取得できます。 定義上、クレードのシーケンスは他のシーケンスよりも互いに類似しているため、このような方法はほとんどのクレードをカバーするはずです。 アルゴリズムは非常に簡単です。サンプルに任意のシーケンスを含め、残りのシーケンスごとに距離を記憶します。 次に、この距離が最大になる残りのサンプルをサンプルに含めます。他のサンプルについては、その距離も覚えています。 次に、残りのものから、サンプル内のシーケンスの1つまでの最小距離が最大のものを選択し、サンプルに追加して、距離を記憶します。 サンプルが目的のサイズに達するまで繰り返します。 近隣結合アルゴリズムに類似しているため、このメソッドを遠隔結合と呼びました。 コードでは 、次のようになります。
leader = not_sampled[0] reduced_list.append(leader) minima = {x: dist(leader, x) for x in not_sampled[1:]} while len(reduced_list) < final_count: leader = max(minima.items(), key=lambda a: a[1]) reduced_list.append(leader[0]) del minima[leader[0]] not_sampled.remove(leader[0]) for i in not_sampled: d = self[(leader[0], i)] if d < minima[i]: minima[i] = d
または画像:
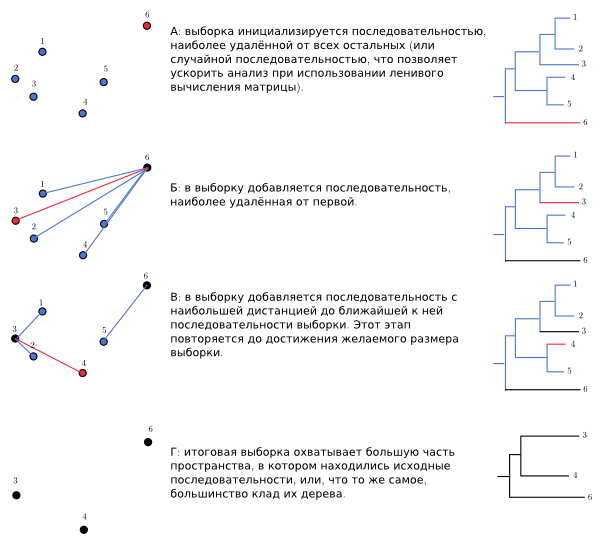
赤-この反復で追加、黒-すでに追加。
判明したように、説明した方法はかなりうまく機能します。 シーケンスの1000のシミュレートされたセットの結果を以下に示します(青-遠い結合、黒-RS、ランダム選択、赤-SS、与えられたものに最も近いシーケンスの数の選択):

シミュレーションごとに、サイズが約12個または2個のトレジャー(平均が15で標準偏差が5の正規分布)が、それぞれ単位から数百のシーケンスのツリーが作成されました。 このツリーによると、ランダムなヌクレオチド配列が生成され、生成されたセットから10%から90%の配列が取得されました。 効率は、少なくとも1つのシーケンスによって削減されたサンプルで表されるクレードの割合として推定されました。 ご覧のとおり、少数のDJシーケンスを選択する場合、他の2つの方法よりもはるかに効果的です。
ところで、どのようなものに最も近いn個のシーケンスのどのような種類の非常に効果的で明らかに無意味な選択ですか? もちろん、これはダウンサンプリングの方法ではなく、アーティファクトの可能性があります。 大まかに言えば、生物学的データベースへのクエリは、「この配列に最も類似する100個の配列を与える」、または「これと少なくとも同じように見えるすべての配列を与える」として定式化できます。 最初の定式化は他のタスクに役立ちます(ほとんどの場合、デフォルトの定式化です)が、系統解析の場合、データセットに重大な不均衡が生じる可能性があります。
実際のデータに基づいて、 AST税法もテストに含めることができます。 以下は、2つのデータセットの結果です。左側は、リボソームRNAの大きなセットです。 これは古典的なマーカー遺伝子であり、重複や削除はほとんど受け継がれないため、種の進化の研究に広く使用されています。 右側には、上のツリーのキチンシンターゼのセットがあります。

結果はかなり予測可能です。rRNAの場合、分類メタデータは進化を完全に記述するため、ASTは他の方法を大幅に回避します。 しかし、彼は上記の理由でキチンシンターゼに対処していません。 上部の水平線はチャートの周囲のフレームではありません。このDJは、データセットが5倍に減少した場合でもすべての宝物をカバーします。
おわりに
厳密に言えば、説明したアルゴリズムには特に生物学的なものはありません。 はい、この場合、シーケンスとそれらの間の距離の生物学的メトリックを使用しました。 ただし、同じ方法で、距離行列があり、サンプルをかなり均等にカバーする必要がある多様性のあるオブジェクトのコレクションからサンプルを取得できます。
Distant Joiningは排出量とは戦わず(これはかなりの多様性と認識されており、サンプルの最初の1つです)、クラスターサイズの比率を維持しないことに注意してください。
Python 3プロトタイプはgithubで入手できます 。 もし彼が突然科学研究の誰かのために重宝するなら、 この記事を引用してください 。