
私は記事が非常に大きいことに気づきました、そして、質問は異なる方向で尋ねられます。 この記事は、ZINBAのインストールに関する質問を別のトピックで収集するために書かれました。 したがって、ZINBAを使用するには、インストール方法を知る必要があります。
詳細なインストールは、開発者のWebサイトに英語で描かれています。 したがって、さらにロシア語で必要な手順を簡単に説明します。 すべての手順はOpenSUSE 12.1 x64 OSで実行されましたが、他のLinuxプラットフォームでは問題なく通過するはずです。 Rを起動し(チューニングに関する質問がある場合はコメントで説明します)、次のコマンドを実行します。
- システム( "wget zinba.googlecode.com/files/zinba_2.01.tar.gz ")。 開発者のサイトからパッケージをダウンロードします。
- install.packages(c(「マルチコア」、「doMC」、「foreach」、「qvalue」、「quantreg」、「R.utils」))。 追加のパッケージをインストールします。 ウィンドウが表示され、ダウンロードするサイトのリストが表示されます。このウィンドウで、これらのパッケージをインストールするサイトを選択します。
- install.packages( "zinba_2.01.tar.gz"、repos = NULL)。 ダウンロードしたパッケージをインストールしています。
- 「Genome Build」セクションの「Necessary file downloads」セクションに進み、使用する1つまたは複数の2ビットゲノムインデックスをダウンロードします。
- 「マッピングファイル」サブセクションで、FASTA / FASTQファイルのように、名前にゲノム名と読み取り長を含むファイルをアップロードします。 ディレクトリに解凍することを忘れないでください。 たとえば、Linuxでは、次のようにダウンロードして解凍できます。「wget www.bios.unc.edu/~nrashid/map##.tgz;tar -xzvf map ##。Tar.gz」。##は長さに対応する番号です。リード。
- Rを開始し、2つのコマンドを順番に実行します。1。generateAlignability(アライメントインデックスを作成する)、2。basealigncount、アライメントの基本的な計算。 このコマンドは、数回の反復で正確なピークマッピングを含むファイルを作成します。
generateAlignability( mapdir=, # , «mappability» outdir=, # , athresh=, # , extension=, # twoBitFile=, # , .2bit ) basealigncount( inputfile=, # , , bowtie outputfile=, # , extension=, # filetype=, # "bed", "bowtie", or "tagAlign" twoBitFile=, # , .2bit )
最初のコマンドは、一度実行してから、エラーの数やフラグメントの長さなどの実験パラメータが変更された場合にのみ実行することをお勧めします。 2番目のコマンドは、実験ごとに実行する必要があり、その後、濃縮された領域とピークの検出品質を向上させる必要がある場合にのみ実行する必要があります(追加の計算負荷が必要になるため)。
これらのアクションの実行中に、多くのエラーを受け取り、テクニカルサポートサービスと話しました。 指示を十分に読んでおらず、彼らでさえもすべてがスムーズではなかったことが判明しました。 最初に、ボウタイを使用してファイルを取得した場合(通常は-Sパラメーターなしの.samではありません)、ファイルの最後の列を削除するだけで、ファイルに7つの列があります。タブだけでなくスペースもセパレーターと見なされます。そのため、フィールドにスペースが含まれていないことを確認してください。 これはバージョン2.01に関連しており、将来修正される予定です。
豊かな地域を見つけるためのチーム自体を以下に説明します。時間がかかり、すべてのリソースを使い果たします。 VirtualBOX仮想マシンでは計算に8時間かかりましたが、4 CPU(石、4pスレッド、1つのCore i7と混同しないでください)、8Gb RAMで、33,000,000回の読み取りからの実験データで実行しました。 8GbのRAMしかない場合、より多くのスレッドをインストールすることはお勧めしません。プログラムは、データよりもページファイルの方が多く動作します。
zinba( refinepeaks=, # ( )? 1 - , 0 - seq=, # input=, # , , . 'none' filetype=, # : 'bed', 'bowtie', 'tagAlign' threshold=, #, p value . , 0.05 align=, # , outdir generateAlignability numProc=, # CPU , ( Core i7 8, 7 , 1) twoBit=, # , .2bit outfile=, # extension=, # ################### ################### basecountfile=, # basecount , refinepeaks 1 broad=, # , TRUE FALSE ( ) printFullOut=, # : 1, ( ); 0, ( ) )
パッケージを作業用に準備しました。今度は、チェックするために何かを開始します。 開発者は、テスト用のファイルセットhttp://www.unc.edu/~nur2/zinbaweb/test_data.tgzを提供しています 。 コマンドラインでは、次のアクションを順番に実行します。
# , . wget http://hgdownload.cse.ucsc.edu/goldenPath/hg19/bigZips/hg19.2bit wget http://www.bios.unc.edu/~nrashid/map36.tgz wget http://www.unc.edu/~nur2/zinbaweb/test_data.tgz # tar -xzvf map36.tgz tar -xzvf test_data.tgz # , , align_athresh1_extension200 mkdir align_athresh1_extension200 # R. R # R library(zinba) generateAlignability( mapdir='map36/', outdir='align_athresh1_extension200/', athresh=1, extension=200, twoBitFile='hg18.2bit' ) basealigncount( inputfile='data/ctcfGM12878rep3chr22.taf', outputfile='data/ctcfGM12878rep3chr22.basecount', extension=200, filetype='tagAlign', twoBitFile='hg18.2bit' ) zinba( align='align_athresh1_extension200/', numProc=4, seq='data/ctcfGM12878rep3chr22.taf', basecountfile='data/ctcfGM12878rep3chr22.basecount', filetype="tagAlign", outfile="data/ctcf", twoBit="hg18.2bit", extension=200, printFullOut=1, refinepeaks=1, broad=F, input='data/inputGM12878rep3chr22.taf' )
データディレクトリには、ctcfという名前と拡張子が.peaks、.peaks.bedのファイルがあります。これらのファイルには、 テーブルの形式で対象のデータが含まれています 。 拡張子が.bedのファイルは、ゲノムブラウザーにダウンロードして表示できます。
NIHウェブサイトwww.ncbi.nlm.nih.gov/geo/roadmap/epigenomics、ftp-trace.ncbi.nlm.nih.gov / sra / sra-instant / reads / ByExp / sra /から取得した他のデータを使用しましたSRX / SRX040 / SRX040388 (ファイルSRR079566.sra)。 以下は、このデータを使用して取得した画像です。 他の入力データでも同様のグラフを取得できます。 赤い線を使用して、写真を2つの部分に分割しました。実験データは上部にあり、標準の注釈(遺伝子、開始部位、イントロン、エクソンなど)は下部にあります。 ZINBAプログラムから結果のベッドグラフだけでなく、ソースデータもダウンロードしました(ソースデータによると、ベッドグラフは記事habrahabr.ru/blogs/bioinformatics/137082に記載されているプログラムを使用して取得されました)。 たとえば、ソースデータにあるいくつかのピークと、ZINBAが検出した濃縮領域を緑色の四角で強調表示しました。 ZINBAから得られたセグメントには増粘が含まれています。 これらの肥厚は、指定されたパラメーターrefignpeaks = 1で計算され、領域の最も濃縮された部分に対応する必要があります。
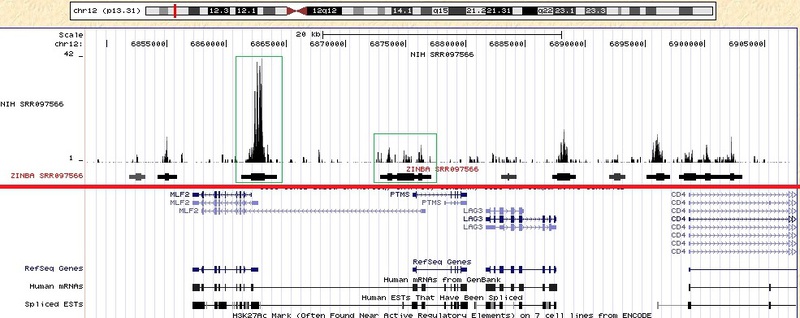
アルゴリズムを徹底的に研究し、他のプログラムの結果と比較した後にのみ、データがどの程度正確かつ確実に処理されるかを理解できます。
レビューは、オハイオ州シンシナティのアンドレイ・カルタショフによって作成されました。porter@ porter.st