sam / bamファイルを操作するには、アセンブルされたsamtoolsパッケージ[1]が必要です。 samtools.sourceforge.netからパッケージをダウンロードし、/ usr / srcなどのディレクトリに展開し、作成したディレクトリに移動してmakeと入力します。 システムにXCursesがインストールされていなかったため、「LIBCURSES = -lXCurses」の行を「LIBCURSES = -lncurses」に置き換えたところ、すべて一致しました。 makeプログラムの結果は、コンパイルされたsamtoolsプログラムとlibbam.aライブラリでした。
読み取り、遺伝子、イントロン、エクソンなどに関する情報を保存するC ++クラスが必要です。 このようなクラスを整理するために、boost.intervalsを使用しましたが、私が望んだ形ではありませんでした。 boost.intervalsもboost.iclも、セグメントに関する完全な情報を保存することはできません。 多くのセグメントに関する次の情報が必要です。
- 開始/終了、開始/長さのペア。
- すべてのセグメントが閉じられています。 両端を含む;
- 特定のポイントで始まるセグメントの数。
- ポイントで交差するセグメントの数。
- 特定のセグメントで始まる、または特定のセグメントと交差するセグメントの数。
特に、この情報は、座標軸の現在のセクションのセグメントによるカバレッジの高さに関する質問に答えます。
新しい記事ごとに、これらのクラスを補充および変更します。 今では、プログラムをまとめて、将来何が欠けているのかを把握するのに十分な数があります。 結果のクラスの例を次に示します。
Reads.hpp
#ifndef _READS_29122011_HPP_ #define _READS_29122011_HPP_ #include <boost/numeric/interval.hpp> #include <QString> #include <QMap> #include <QtDebug> namespace genome { namespace bni = boost::numeric; typedef bni::interval<int> read_position; /********************************************************************************** **********************************************************************************/ class Read { public: Read(): multiplying(1), length(0){}; Read(Read const &r): multiplying(r.multiplying), length(r.length), position(r.position){ sentenceRepresentation=r.sentenceRepresentation; qualityRepresentation=r.qualityRepresentation;}; Read(int start,int len,QString sr="",QString qr=""): multiplying(1), length(len), position(start,start+len-1), sentenceRepresentation(sr), qualityRepresentation(qr){}; int getLevel() {return multiplying;}; void plusLevel() {++multiplying;}; int getStart() {return position.lower();}; int getLength() {return length;}; void operator+= (const int& c) {this->multiplying+=c;}; bool operator== (const Read& r) const {return this->position==r.position;}; bool operator!= (const Read& r) const {return this->position!=r.position;}; void operator++ (int) {this->multiplying++;}; private: int multiplying; int length; read_position position; QString sentenceRepresentation; QString qualityRepresentation; }; /********************************************************************************** **********************************************************************************/ typedef QMap<int,Read> cover_map; class Cover { public: Cover():max_len(0){}; void add(Read&); int getHeight(int); int getHeight(int,int); int getStarts(int); int getStarts(int,int); QList<int> getStarts(); cover_map::iterator getBeginIterator(){return covering.begin();}; cover_map::iterator getEndIterator(){return covering.end();}; bool operator== (const Cover& c) const {return this==&c;}; bool isEmpty(){return covering.size()==0;}; // static Cover empty(){ return Cover();}; private: cover_map covering; int max_len; }; /********************************************************************************** **********************************************************************************/ class Lines { public: Lines(){}; Lines(Lines&){}; void addLine(QString, Read&); Cover& getLineCover(QString); QList<QString> getLines(void) { return lines.keys(); }; /* */ private: QMap<QString,Cover> lines; }; /********************************************************************************** **********************************************************************************/ class GenomeDescription: public Lines { public: quint64 notAligned; // number of reads (ussualy form sam/bam file) that are not aligned quint64 total; /* */ void setGene(const QChar &sense,const QString &chrName,const qint32 &pos,const qint32 &,const qint32 &len) { Read r(pos,len); addLine(chrName+sense,r); }; /* */ GenomeDescription():Lines(), notAligned(0), total(0) {}; }; } #endif
では、samファイルを操作するための関数のラップを開始しましょう。 samとbamの違いは、最初のファイルはフォーマットされているもののプレーンテキストファイルであり、2番目のファイルは圧縮されたバイナリファイルであるということです。 構造化。 多数の読み取りを行うbamを使用することを好みます。samファイルはギガバイトに達する可能性があります。
BEDHandler.hpp
#ifndef BEDHandler_H #define BEDHandler_H #include <config.hpp> #include <Reads.hpp> template <class Storage, class Result> class BEDHandler : public QState { private: Storage *sam_input; Result *output; QSettings setup; QFile _outFile; public: BEDHandler(Storage &sam,Result &output,QState *parent=0); ~BEDHandler(); protected: virtual void onEntry(QEvent* event); }; #include <BEDHandler.cpp> #endif
BEDHandler.cpp
//------------------------------------------------------------- //------------------------------------------------------------- template <class Storage,class Result> BEDHandler<Storage,Result>::~BEDHandler() { } //------------------------------------------------------------- //------------------------------------------------------------- template <class Storage,class Result> BEDHandler<Storage,Result>::BEDHandler(Storage& sam,Result &_output,QState * parent): QState(parent), sam_input(&sam), output(&_output) { _outFile.setFileName(setup.value("outFileName").toString()); _outFile.open(QIODevice::WriteOnly|QIODevice::Truncate); } //------------------------------------------------------------- //------------------------------------------------------------- template <class Storage,class Result> void BEDHandler<Storage,Result>::onEntry(QEvent*) { if(!setup.contains("graphWindow")) setup.setValue("graphWindow",200); if(!setup.contains("siteShift")) setup.setValue("siteShift",75); if(!setup.contains("separateStrand")) setup.setValue("separateStrand",false); if(!setup.contains("HeaderString")) setup.setValue("HeaderString","track type=bedGraph name=%1"); if(setup.value("HeaderString").toString()!="") { _outFile.write((setup.value("HeaderString").toString().arg(_outFile.fileName())+"\n").toLocal8Bit()); _outFile.flush(); } quint32 window=setup.value("graphWindow").toUInt(); quint32 shift= setup.value("siteShift").toUInt(); QString line; QString chrome; foreach(line,sam_input->getLines()) { if(line.endsWith("-")) continue; chrome=line; chrome.truncate(line.length()-1); QMap <int,int> bed; { genome::cover_map::iterator i=sam_input->getLineCover(chrome+QChar('+')).getBeginIterator(); genome::cover_map::iterator e=sam_input->getLineCover(chrome+QChar('+')).getEndIterator(); while(i!=e) { int val=i.key()+shift; bed[val-val%window]+=i.value().getLevel(); ++i; } } { genome::cover_map::iterator i=sam_input->getLineCover(chrome+QChar('-')).getBeginIterator(); genome::cover_map::iterator e=sam_input->getLineCover(chrome+QChar('-')).getEndIterator(); while(i!=e) { int val=i.key()-shift; if(val<0) val=0; bed[val-val%window]+=i.value().getLevel(); ++i; } } QMap<int,int>::iterator i = bed.begin(); for(;i!=bed.end();i++) { _outFile.write(QString(chrome+"\t%1\t%2\t%3\n"). arg(i.key()).arg(i.key()+window).arg(i.value()).toLocal8Bit()); _outFile.flush(); } } }//end of function
記事の残りのコードは提供しません。 アーカイブへのリンクです 。構造は非常に単純です。ルートには2つのサードパーティディレクトリとsrcディレクトリがあり、最初にsamtools、2番目にsam2bedgraphとglobalがあります。 プロジェクトをビルドするには、sam2bedgraphディレクトリでqmakeを実行してからmakeを実行する必要があります。 ネイティブQt(4.7.4)およびブースト(1.46.1)を使用したopenSUSE 12.1 x64でチェック。
ベッドグラフファイル構造の説明へのリンクは最初の段落にありますが、簡単に説明します。 最初の行は、ファイルの特性を設定します(存在する場合)。 ファイルの本文には、少なくとも4列が含まれています。 最初の列には、染色体の名前が示され、2番目と3番目に-セグメント(「ウィンドウ」)の開始と終了の座標がそれぞれ示されます。 最後の列-「高さ」、この場合、これはこのウィンドウの読み取り数です。 各ファイルの「ウィンドウ」のサイズは、実験的に選択する方が適切です。 ChIP-seqには200 bpウィンドウを使用し、RNA-seqには20 bpウィンドウを使用します。
プログラムには、入力.bamファイルと、bedgraphに関するデータが書き込まれる出力ファイルの2つのパラメーターがあります。 パラメーターが設定されていない場合、プログラムはデフォルトのファイルinput.bamを開き、出力output.dataを作成しようとします。 構成ファイル.config / CCHMC / sam2bedgraph.iniを使用して、ユーザーのホームディレクトリにディレクトリが作成されます。このファイルでは、デフォルトのファイル名の値を変更し、「ウィンドウ」の長さを変更できます。このウィンドウの高さを考慮し、最初の情報行の外観を設定します
結果のファイルは、ゲノムブラウザでダウンロードできます。 Genome.ucsc.edu [2]サイトにアクセスし、 [Genomes]リンクをクリックします。
次のページに進み、bamファイルを受信する条件を設定する必要があります。クレード(進化ライン)、ゲノム(ゲノム)、アセンブリ(要約、アセンブリ)です。 そして、「カスタムトラックを追加」ボタンをクリックします。
「ファイルを選択」ボタンをクリックし、「送信」ボタンをクリックしてファイルをダウンロードできます。 その結果、注釈付きのページが表示され、データベースとベッドグラフからの情報が表示されます。 ゲノムブラウザーをローカルにインストールする場合、セッション内で一時的にではなく、常にベッドグラフをサイトに追加できます。 サイトへの特別なリンクを整理して、bedgraphファイルの入手先を示すことができます。 このサイトでは、ゲノムブラウザーのコピー方法について説明しています。
複数のファイルをアップロードできますが、私はそれを行いました。 次のスクリーンショットに結果が表示されます。
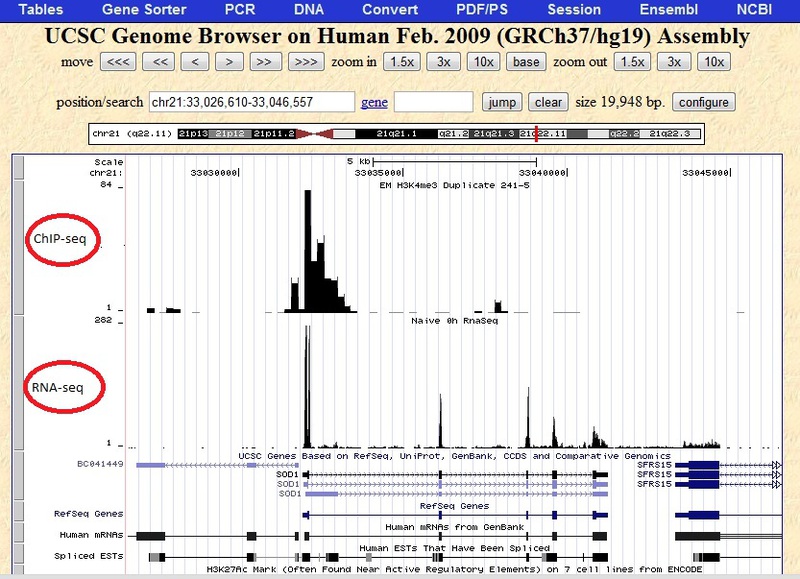
このスクリーンショットでわかるように、レベルがゼロの領域があり、ピークのある濃縮された領域があります。 背景やノイズが見える場合があります。 また、ChIP-seqがRNA-seqとどのように異なるかがはっきりとわかります。 プログラムからのこれらの領域の定義は別の質問であり、単一の答えはありません。 事実は、ゲノム内の遺伝子と同じ数のピークが存在する可能性があり、さらに悪いことには、いくつのエクソンが存在する可能性があるということです。 そして、実験の枠組みの中で豊かな領域を分離することは非常に面倒です。
1. Li、H.、et al。、The Sequence Alignment / Map format and SAMtools。 バイオインフォマティクス、2009.25(16):p。 2078-9。
2.ケント、WJ、他、UCSCのヒトゲノムブラウザ。 Genome Res、2002.12(6):p。 996-1006。
3. Dreszer、TR、et al。、UCSC Genome Browser database:extensions and updates 2011. Nucleic Acids Res、2012. 40(データベースの問題):p。 D918-23。