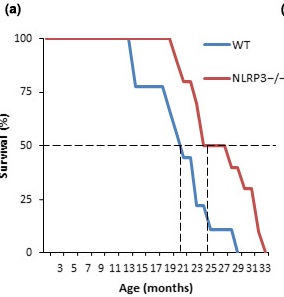
The whole study can be told in two sentences: scientists created a mouse line in which the NLRP3 gene is knocked out. As a result of this, the mice lived about 30% longer than the control, and remained healthier. Here's a look at the survival graph. WT, the blue line is wild type, wild-type mice, NLRP3 - / -, the red line are mice with the knocked out NLRP3 gene. True, we certainly did not understand, but there were about 60 mice in total, but this is not accurate. They wrote to the authors to find out for sure.
And here we have to think a little. Scientists have finally found an extra gene, and if this gene is cut down, can mice extend life by a few percent? Maybe I also cut down this gene for myself?
The inner skeptic tells us that it’s too early to rejoice. And indeed: the mice lived under conditions called “specific pathogen ‐ free conditions” —that is, conditions free of any selected pathogens. That is, such pathogens that ruin the experiment for scientists.
The fact is, the NLRP3 gene product is the cryopyrin protein, which is important for the innate immune response. Cryopyrine is a cytosolic protein, Nod-like receptor of the NALP family, the main component of the same type of inflammasome.
Inflammasoma is such a multi-protein complex responsible for the activation of the inflammatory response. Promotes the secretion of pro-inflammatory cytokines: IL-1β and IL-18. And their secretion, in turn, causes pyroptosis - this is a type of programmed cell death in which the cell membrane breaks and all its contents are poured out.
Here pyroptosis is the very important process of innate immunity that we talked about. Pyroptosis limits the reproduction of intracellular pathogens.
Now we need to explain how the NLRP3 gene is associated with cardiovascular disease. And so: the level of associated with it increases with mylamocardial infarction, with atherosclerosis, coronary heart disease, diabetic cardiomyopathy, chronic heart failure and hypertension (Bullón et al., 2017; Liu, Zeng, Li, Mehta, & Wang, 2017) . Well, in general, markers of inflammation are associated with cardiovascular disease.
It has already been shown that a genetic deletion (loss of a chromosome with a gene) of NLRP3 in mice improves their health by attenuating multiple age-related degenerative changes (Youm, 2013). Moreover, NLRP3 in old mice increased muscle strength and stamina and prevented an age-related increase in the number of myopathic fibers (McBride et al., 2017).
But the role of NLRP3 inflammation in longevity and aging of the heart has not been studied. And in the work we are talking about here, scientists set the task of determining whether the NLRP3 gene knockout can affect life expectancy and potentially prevent heart aging.
To do this, they ran an experiment in which they measured many different markers for mice. Honestly, the list of these markers made me deeply satisfied, because I would have measured it myself.
These are the markers:
- fasting glucose tolerance. Mice fasted at night for 16 hours, and then they injected glucose into the abdominal cavity at a rate of 1 g / kg;
- leptin, adiponectin, IGF ‐ 1 in serum;
- serum biomarkers: glucose, triglycerides, cholesterol, uric acid, aspartate aminotransferase, alanine aminotransferase and creatine kinase;
- electrocardiography.
And here is what they did:
- In NLRP3 - / - mice (with the NLRP3 gene disabled), the average life expectancy was increased by 34% compared to the control group of animals and the maximum life expectancy was 29%.
- Control mice by 24 months were more bald than experimental.
- Experimental were more glucose tolerant.
- Blood glucose and circulating IGF ‐ 1 levels were reduced in young and old NLRP3 - / - mice, indicating that their insulin sensitivity was higher than that of the control.
- Leptin in young and old NLRP3 - / - was approximately at the same level as in the control, but the ratio of leptin to adiponectin with an increased level of adiponectin was lower in old NLRP3 - / - mice. Leptin is a weight regulator, has not changed, and okay. The mass of control and experimental mice and the amount of food consumed by them did not differ either. But an imbalance in the ratio of leptin to adiponectin is associated with CVD, metabolic syndrome and non-alcoholic fatty liver disease.
- Plasma lipids were lower in NLRP3 - / - old mice.
- Active caspase 1 and IL-1β levels were higher in older control mice than in NLRP3 - / - mice.
- Elevated levels of TNF-α, IL-6, and IL-8 were observed approximately the same in both control and NLRP3 - / - mice. This shows that the loss of NLRP3 did not affect the age-related increase in other inflammatory pathways.
- Heart mass was higher in wild control mice compared to NLRP3 - / - mice. Heart hypertrophy, measured by the thickness of the wall of the left ventricle, was significantly increased in old wild mice compared to NLRP3 - / - mice
Electron microscopic analysis showed that the number of accumulated autophagosomes was reduced in the hearts of old NLRP3 - / - mice. This is because the inhibition of NLRP3 caused an improvement in the quality of autophagy in the heart during aging. The data obtained show that the knockout of the NLRP3 gene prevented many age-related changes in the heart, retained heart function in old mice, increasing life expectancy.
Here is another important detail. In the Discussions section, the authors say that an increase in glucose tolerance, a decrease in the ratio of leptin and adiponectin, as well as regulation of dyslipemia are associated with common pathways such as IGF-1, PI3K / AKT / mTOR, and autophagy and intracellular levels of NAD + are also involved. Older NLRP3 - / - mice had low serum levels of IGF-1. The role of IGF-1 is contradictory, but the authors of the work write that low levels of IGF-1 in serum are the final product of a decrease in insulin / IGF-1 signaling, which is known to prolong life in both invertebrates and vertebrates (Finkel, 2015 )
What does this work teach us? Which of it can be concluded?
The authors have measured a lot of things, so the conclusions may be different.
For example: inflammation plays a large role in the development of CVD, but not anyhow, but one in which pyroptosis is involved. Indeed, TNF-α, IL-6, and IL-8 did not differ in control and experimental mice.
Or this: everything depends on insulin and IGF-1.
Or this: in any incomprehensible situation, measure as many markers as possible, even if it seems to you that pyroptosis, glucose tolerance, and the leptin / adiponectin ratio have nothing in common. Just take the biomarker panel and measure as much as possible, the result will be interesting.
Or this: it’s always like that with genes - you’ll chop off one, the whole system will go. And in order to figure out where she’s going, you must make gene maps, fill out databases and study how these genes relate. No, it's true: who would have thought that pyroptosis, that is, the ability of a cell to burst and spill its contents into the nearby intercellular matrix, is somehow related to aging, and even through glucose?
Here, let's take a closer look. Genes do not work alone. They are all tied to each other there, one regulates the other, the butterfly effect. And the ambush is that we see very poorly what affects what. We poke around like blind kittens, hoping to find the right therapeutic targets. We need a map, guided by which, we could easily find a way from point A to point B. B is immortality))
That is why we began to make our base of all these genes associated with increased life expectancy. We will talk about it later, possibly in December.
Our task is to establish many interconnections that play a decisive role in the possible increase in human life expectancy. Plus, specifically this article caused a lot of discussion in the Open Longevity community about whether it is possible to reproduce its results. Therefore, it is also good to have a base on the genes of aging and longevity, evaluating the reliability of scientific results.
Here is the full text of the article that we discussed here for thought.
D. Bataeva, A. Rzheshevsky, M. Batin.