 バイオインフォマティクスに関する最初のレビュー記事を公開してからほぼ1年が経過しました 。 もっと頻繁に書きたいのですが、バイオインフォマティクスだけが気を散らし、許可していません。 前の記事で、ゲノムを組み立てるタスクと、それらが関与しているアルゴリズム生物学研究所( SPbAU RAS )について言及しました。 すでに述べたように、世界中の多くの大学がこのタスクに長い間取り組んでおり、非常に成功しています。 しかし、ゲノムを組み立てるために、生物学者は膨大な量のさまざまな種類のデータを取得します。それぞれが独自の特性を持っています。 約5年前、 MDAテクノロジーが登場し、バクテリアの研究に大きな機会が開かれました。 その結果、新しいゲノムコレクターが必要なデータタイプが作成されました。 数年後、スタンフォード大学やマサチューセッツ州ではなく、サンクトペテルブルクの若いアカデミック大学で開発されました。 しかし、まず最初に。
バイオインフォマティクスに関する最初のレビュー記事を公開してからほぼ1年が経過しました 。 もっと頻繁に書きたいのですが、バイオインフォマティクスだけが気を散らし、許可していません。 前の記事で、ゲノムを組み立てるタスクと、それらが関与しているアルゴリズム生物学研究所( SPbAU RAS )について言及しました。 すでに述べたように、世界中の多くの大学がこのタスクに長い間取り組んでおり、非常に成功しています。 しかし、ゲノムを組み立てるために、生物学者は膨大な量のさまざまな種類のデータを取得します。それぞれが独自の特性を持っています。 約5年前、 MDAテクノロジーが登場し、バクテリアの研究に大きな機会が開かれました。 その結果、新しいゲノムコレクターが必要なデータタイプが作成されました。 数年後、スタンフォード大学やマサチューセッツ州ではなく、サンクトペテルブルクの若いアカデミック大学で開発されました。 しかし、まず最初に。
再びゲノムアセンブリー
基本的な概念を簡単に思い出してください。 生物学者のゲノムは、4種類のヌクレオチド(アデニン、シトシン、グアニン、およびチミン)の二重鎖である長いDNA分子です。 たとえば、細菌のDNA鎖の長さは数百万のヌクレオチドで測定されますが、人間のDNAの長さは約30億です。 バイオインフォマティクスの場合、ゲノムは4文字のアルファベット{A、C、G、T}の上にある大きな線です。 DNA分子全体を「読む」ことは完全に不可能です。 最新の技術では、ランダムな場所から数百ヌクレオチドの小さな断片のみを読み取ることができ、それでもエラーが発生します。 これらのピースは読み取りと呼ばれ、読み取りプロセスはシーケンスと呼ばれます。 ある実験では、数千または数億の測定値が作成されます。 次に、ゲノムアセンブラー、これらの読み取り値から元のシーケンスを復元しようとするプログラムを開発するバイオインフォマティクスが登場します。 原則として、アセンブラーはゲノム全体を回復することはできませんが、数百万の短い読み取りから数十の配列を数十万のヌクレオチド長にすることができます。 このようなシーケンスは、生物学者によってすでにさらに研究されています。
 誰がゲノムを収集して分析する必要がありますか? たとえば、ヒトゲノムのアセンブリは、初期段階で体内のがん細胞の存在を判断するのに役立ちます。 もう1つの目標は、いわゆる個別化医療への移行です。 各人が遺伝子を知っている場合、投薬および治療は症状だけでなく、彼の遺伝的特徴も考慮して処方することができます。 個人医療はまだ普及していませんが、一部の診療所では、患者のゲノムの配列決定のためのサービスを既に提供しています。 ゲノムの読み取りコストは常に低下しており、新しいアセンブリアルゴリズムにより、ゲノムをより速く実装できます。 それにもかかわらず、ヒトゲノムだけでは完全な診断には十分ではありません。 私たちのそれぞれには、膨大な数の内部プロセスに関与し、生命に必要なさまざまな細菌の数千種がいます。 これらの細菌のゲノムの集合(その全長がヒトゲノムの長さを超える)は、人体のより良い研究を可能にするでしょう。 ただし、シーケンスには困難があります。
誰がゲノムを収集して分析する必要がありますか? たとえば、ヒトゲノムのアセンブリは、初期段階で体内のがん細胞の存在を判断するのに役立ちます。 もう1つの目標は、いわゆる個別化医療への移行です。 各人が遺伝子を知っている場合、投薬および治療は症状だけでなく、彼の遺伝的特徴も考慮して処方することができます。 個人医療はまだ普及していませんが、一部の診療所では、患者のゲノムの配列決定のためのサービスを既に提供しています。 ゲノムの読み取りコストは常に低下しており、新しいアセンブリアルゴリズムにより、ゲノムをより速く実装できます。 それにもかかわらず、ヒトゲノムだけでは完全な診断には十分ではありません。 私たちのそれぞれには、膨大な数の内部プロセスに関与し、生命に必要なさまざまな細菌の数千種がいます。 これらの細菌のゲノムの集合(その全長がヒトゲノムの長さを超える)は、人体のより良い研究を可能にするでしょう。 ただし、シーケンスには困難があります。
人間から細菌へ
ゲノムの配列を決定するには、十分な量の遺伝物質、つまり同じDNA分子が必要です。 標準的な実験には、数千または数億の同一の細胞が必要です。 人間を含む多細胞生物の場合、十分な量の遺伝物質が数グラムの唾液に含まれているため、これは問題ではありません。 同じ量の細菌細胞を取得することはより困難です-それらは実験室で成長しなければなりません。 問題は、すべての細菌を培養できるわけではないということです。 たとえば、人間の腸内の細菌は大きなコロニーに住んでおり、互いに別々に存在して増殖することはできません。 コロニーから採取した1つの細菌を培養することは不可能です。
今日、コロニー内の未培養細菌をシーケンスするための2つの根本的に異なるアプローチがあります。 最初の、バイオテクノロジーの観点からはかなり単純な、メタゲノムです。 それは次のもので構成されています。コロニーを取得し、すべての細菌からDNA分子を選択し、それらをシーケンサーに入れて読み取ります。 その結果、数十種類の異なるバクテリアのゲノムの読み取り値を同時に受信します。 密接に関連する細菌では、ゲノムは互いに類似しており、異なる種類の細菌では非常に異なっていますが、同一の領域を含んでいる場合があります。 これらの要因は、この種のデータの収集を非常に複雑にします。 今日行われ、現在進行中のメタゲノム研究の数にもかかわらず、そのようなデータに基づいて生物学者を満足させる結果を生み出すアセンブラーが少なくとも1人いるとは言えません。 原則として、コレクターは多数のエラーを含む短いシーケンスを生成し、分析が困難です。 時には、メタゲノムプロジェクトでは、さまざまな細菌のセット全体に対するいくつかの遺伝子の発見がすでに成功していることがあります。
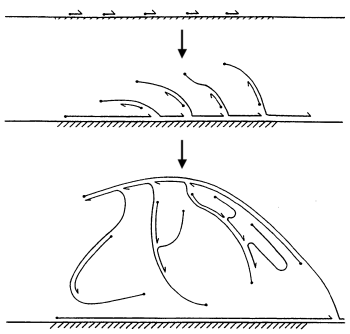 培養されていない細菌をシーケンスするための別のアプローチは、ほんの数年前に発見され、かなり複雑なバイオテクノロジーのプロセスで構成されています。 コロニーから興味のあるバクテリアを1つ取り出し、そこからDNAを抽出し、DNAにいくつかの「コピーマシン」を植えます。これらのコピーマシンは、分子の周りを「走り」、そのセクションをランダムにコピーし始めます。 コピーするとき、新しいDNA鎖は分子から分岐します。これは、「コピーマシン」に取り込まれ、遺伝物質を蓄積し続けます。 結果として、興味のあるDNAのセクションの同一のコピーを取得し、それをシーケンサーに入れます。 単一細胞ゲノムをシーケンスするこの方法は、Multiple Displacement Amplification(MDA)と呼ばれます。 すべてが素晴らしいようです。 しかし、悲しいかな、問題があります。 「コピー機」は単独で動作し、制御できません。 したがって、DNAの一部のセクションは1万回コピーできますが、他のセクションは決してコピーできません。 その結果、単に材料が不足しているために、一部の地域がアセンブリに存在しなくなります。 この方法の結果として、平均して、その構造(一部はコピーに悪影響を及ぼす)と実験の質に応じて、ゲノムの95〜99%を復元することが可能です。
培養されていない細菌をシーケンスするための別のアプローチは、ほんの数年前に発見され、かなり複雑なバイオテクノロジーのプロセスで構成されています。 コロニーから興味のあるバクテリアを1つ取り出し、そこからDNAを抽出し、DNAにいくつかの「コピーマシン」を植えます。これらのコピーマシンは、分子の周りを「走り」、そのセクションをランダムにコピーし始めます。 コピーするとき、新しいDNA鎖は分子から分岐します。これは、「コピーマシン」に取り込まれ、遺伝物質を蓄積し続けます。 結果として、興味のあるDNAのセクションの同一のコピーを取得し、それをシーケンサーに入れます。 単一細胞ゲノムをシーケンスするこの方法は、Multiple Displacement Amplification(MDA)と呼ばれます。 すべてが素晴らしいようです。 しかし、悲しいかな、問題があります。 「コピー機」は単独で動作し、制御できません。 したがって、DNAの一部のセクションは1万回コピーできますが、他のセクションは決してコピーできません。 その結果、単に材料が不足しているために、一部の地域がアセンブリに存在しなくなります。 この方法の結果として、平均して、その構造(一部はコピーに悪影響を及ぼす)と実験の質に応じて、ゲノムの95〜99%を復元することが可能です。
MDAテクノロジーの問題
シーケンス中にゲノム内のヌクレオチドが読み取られる回数は、そのコーティングと呼ばれます。 標準シーケンスでは、すべてのヌクレオチドのカバー率はほぼ同じです(下図のコーティングプロットの例)。 MDAテクノロジを使用する場合、非常に不均一なカバレッジのデータを取得します(下の右図)。これは、アセンブリタスクにとって重大な問題です。 理由を理解してみましょう。
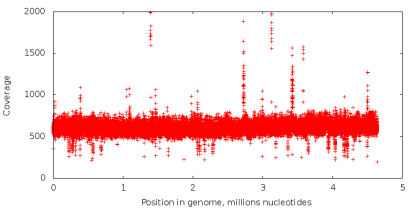
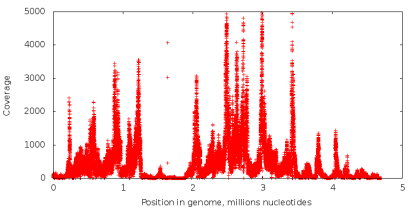
以下の実験を実施します。 標準的なシーケンスを使用して取得したデータを取得し、固定長kのすべての読み取り部分文字列から抽出します(以降-kメジャー) :ACG、CGT、GTA、およびTAC)。 次に、読み取り値で各kメジャーが何回発生するかを計算します。 特定のkメジャーが十分な回数出現する(つまり、カバレッジが高い)場合、それが元のゲノムにも発生していると仮定する理由があります。 k-merがまれな場合は、おそらく誤って読み取られたヌクレオチドが含まれています。 シーケンスエラーはランダムなイベントであり、多数のkメジャーで同じエラーがすぐに発生する可能性は非常に小さいです(単一ヌクレオチドの誤った読み取りの確率は、読み取りのヌクレオチドの位置に応じて0.001%から1%の範囲です)。 したがって、kメジャーのカバレッジ値を使用して、それらを信頼性の高いエラーと誤ったエラーに分割できます。 同様の方法は、最新のアセンブラとユーティリティで読み取り値のエラーを修正するために使用されます。 ただし、単一セルシーケンスの場合は、この方法でkメジャーを分離することはできません。これは、まれなkメジャーがコピーされる領域に少数回対応し、エラーがまったく含まれないためです。
MDAアプローチの問題は、不均一なカバレッジだけではありません。 DNAをコピーすると、新しい一本鎖が主鎖から分岐します。 元のゲノムの完全に異なる部分に対応するDNA配列が近くにあるため、別の出芽鎖とランダムにくっつくことができます。 このような誤った接着の代わりに、「コピー機」が再び座って間違ったセクションを伝播する可能性があります。 次に、シーケンスの結果として、ゲノムのさまざまな部分からの配列を含む読み取り値(キメラ読み取り値)を取得します。 このような読み取り値を処理する際、ゲノムコレクターは2つのシーケンスを誤って1つに結合する場合があります。 誤って配置された1つのヌクレオチドが重大なエラーではない場合、2つの誤って接着されたサイトははるかに深刻な問題であり、さらなる分析を複雑にします。
最後に
サンクトペテルブルクのアルゴリズム生物学研究所での研究の主要な主題となったのは、MDAテクノロジーを使用して得られたデータです。 不均一なカバレッジとキメラ読み取りの問題を回避する多くのアルゴリズムが開発されました。 これらのアルゴリズムは、ゲノムアセンブラーSPAdesに実装されました。これは、今日では細菌ゲノムのアセンブリーのリーダーであり、世界の主要な研究所で使用されています(たとえば、細菌ゲノム研究の世界最大のセンターである米国のJoint Genomic Instituteで)。
さらに、現代の標準アセンブラーの基本原則を読者に喜んで紹介します。単一のセルで取得したデータを収集するために使用される新しいアプローチとアルゴリズムについて話しますが、残念ながら、これは1つの記事に収まりません。 アセンブリアルゴリズムについて詳しく知りたい場合は、このトピックに関する一般的なテキストや科学出版物へのリンクを以下に示します。 次の記事が今回よりも早く登場することを心から願っています。
興味のある方へ
- 「コピー機」の機能は、ランダムな場所でDNAの一部を複製できるタンパク質であるPhi-29 DNAポリメラーゼによって実行されます。
- MDAを使用して得られたデータのシーケンスエラーを決定および排除するために、たとえばハミング距離にわたるkメジャーのクラスタリングに基づくアプローチ、エラーのある場所でのde de Bruyneのトポロジカルな特徴が使用されます。
- キメラ化合物の検索と除去も、主にカウント・ド・ブラインのトポロジーに従って実行されます。
参照資料
- PEC CompeauおよびPA Pevzner。 ゲノム再構築:10億個のパズル ( ゲノムアセンブリレビュー)
- PA Pevzner、H。Tang、およびM. Waterman。 DNAフラグメントアセンブリへのオイラーパスアプローチ。 アメリカ合衆国国立科学アカデミーの議事録、98:9748-9753、2001 (ゲノムアセンブリのタスクでのデブリュイン伯爵の使用に関する最初の記事)
- D.ゼルビーノ、E。バーニー。 Velvet:de Bruijnグラフを使用したde novoショートリードアセンブリのアルゴリズム。 ゲノム研究。 18(2008)、821–829 (クラシックアセンブラーベルベット)
- A.バンクビッチ、S。ヌルク、他 SPAdes:新しいゲノムアセンブリアルゴリズムとそのシングルセルシーケンスへの応用。 Journal of Computational Biology 19(5)(2012)、455-477 (SPAdes-MDAを使用して取得したデータをアセンブルするためのアセンブラー)