 2013年の記事「次世代シーケンスデータからの単一ヌクレオチド多型の識別のためのサポートベクターマシン」 (O'Fallon、Wooderchak-Donahue、Crockett)は、サポートベクター法 (SVM)を使用してゲノムの多型を決定する新しい方法を提案しています) 2011年の記事「次世代DNAシーケンスデータを使用した変異の発見とジェノタイピングのフレームワーク」では 、機械学習法を使用して単一ヌクレオチド多型 ( SNP 、スナップ)を決定することについて既に説明しましたが、SVMの使用に基づくアプローチが説明されていますこの記事で初めて。
2013年の記事「次世代シーケンスデータからの単一ヌクレオチド多型の識別のためのサポートベクターマシン」 (O'Fallon、Wooderchak-Donahue、Crockett)は、サポートベクター法 (SVM)を使用してゲノムの多型を決定する新しい方法を提案しています) 2011年の記事「次世代DNAシーケンスデータを使用した変異の発見とジェノタイピングのフレームワーク」では 、機械学習法を使用して単一ヌクレオチド多型 ( SNP 、スナップ)を決定することについて既に説明しましたが、SVMの使用に基づくアプローチが説明されていますこの記事で初めて。
ゲノムの多型の決定は重要です(たとえば、 GWAS協会の ゲノム全体の検索など )が、重要な作業です。 多くの生物はヘテロ接合体であり、データに誤った情報が含まれている可能性があることに留意する必要があります。
SNPを決定するための標準的なアプローチは、参照(参照)ゲノムに関する配列決定データ(読み取り、ゲノムの断片)のアライメントに基づいています。 ただし、読み取りにはエラーが含まれる場合があり、参照用に正しく整列されない場合があります。 この位置でのエラーの発生は、多型の出現よりもまれなイベントであると考えられているため、この場合の最も簡単なアプローチは、低頻度のバリアントを除外することです。 ただし、2010年の記事「全ゲノムシーケンスによる一般的な疾患のまれな変異体の役割の解明」では、1%未満の頻度で発生するまれな多型が多くの疾患に最も大きな影響を与えることが示されました。 頻度ベースのフィルタリングは、まれなSNPに対して多数の偽陰性を伴うため、ゲノム変異を決定するためのより感度の高い方法を作成する必要があります。
記事で示されているように、機械学習に基づく方法の利点は、ゲノムの特定の位置での多型の可能性に影響するさまざまな要因を組み合わせることができることです。これにより、よりまれな多型に対する方法の感度も向上します。
他の機械学習方法(たとえば、 上記の記事で説明した強化学習アプローチ)とは異なり、SVMは必要なトレーニングデータが大幅に少なくなります。
この記事では、アルゴリズムを実装するための詳細な方法については説明しませんが、技術的な詳細を含む他の記事へのリンクを提供します。 SVMアルゴリズムの実装を提供するプラットフォームとして、 LIBSVMが使用されます。 LIBSVMに関する最初の出版物は2011年にさかのぼり、サイトの最後のリリースは2013年を参照しています。SAMまたはBAMファイルは入力として受け入れられます。これは、バイオインフォマティクスへの便利で自然なアプローチのようです。 アルゴリズムの結果として、 VCFファイルが生成されます。
モデルのトレーニングには2種類のデータが使用されます。
- 真の多型に対応するアライメント。
- 多型の存在に関連しない特定の位置でのヌクレオチドのミスマッチ。
2番目のタイプに対応するデータをどのように収集できるのでしょうか。 著者は、例として2つの可能な戦略を引用しています。
- UnifiedGenotyperなど、 GATKのベストプラクティスガイドラインを使用して多型を特定します。 著者は、1つのケース( 1000 Genomesプロジェクトデータに記載されているスナップを除く)でのみ定義されたスナップを使用し、残りのスナップは、非常に低レベルのアライメント品質のスナップを選択しました。
- 著者らは、57 個のエクソームのローカルデータベースで少なくとも8回発生する多型を選択しました (1000個のゲノムからのデータにも対応していません)。
多型はゲノムの転写されていない領域でも発生する可能性があるため、エクソムの分析のみから得られた情報を使用すると、一部の不正確さが生じる可能性があることに注意してください。
得られたデータに基づいて、トレーニングが実行されます。 この記事には、トレーニングには、トレーニングが実行される2つのクラスのデータを最もよく分離する超平面の数値パラメーターの決定が含まれていると記載されています。
また、著者がモデルのトレーニングのために選択した機能は興味深いものです。 著者は、3つの属性で構成されるセットから始めてモデルをトレーニングしました。 これは悪い結果をもたらしたので、多くの兆候が拡大しました。 このプロセスの詳細はこの記事では説明していません。また、著者が新しい機能を追加した順序は完全には明確ではありません。 合計で、著者は16の兆候を調べました。そのうち、読み取りのエラーの確率、読み取りのヌクレオチドの平均品質、この位置のヌクレオチドの品質の合計(最後の2つは多くの記号に追加しても改善しませんでした)、読み取りのアライメントの品質、シーケンシングの深さ(最後のまた、モデルを改善しなかった)、対立遺伝子のバランス、隣接ヌクレオチドの品質。
記事で与えられたグラフによると、説明された方法は、特定のデータセットNA12878の感度と特異性の多型を決定する他の方法よりも優れています。 このメソッドは、すべての真陽性の95.6%を決定しましたが、他のメソッド:UnifiedGenotyper、UnifiedGenotyper + VQSR 、 SAMtoolsおよびFreeBayesは 、それぞれ90.6%、94.1%、89.5%および88.7%のみを決定しました。
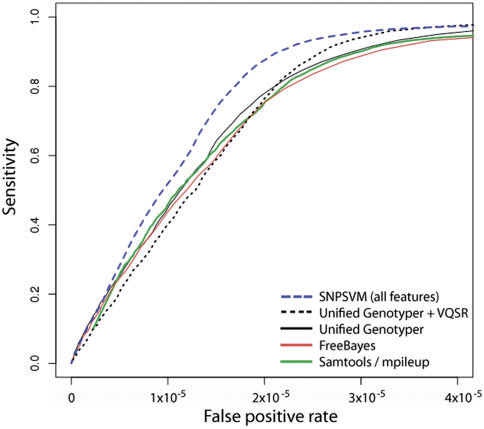
説明されたアプローチの欠点の中で、SVMモデルは異なるエラープロファイルを含むデータに対して悪い結果を与える可能性があることに注意することができます。 さらに、マルチスレッドにもかかわらず、実装はGATK UnifiedGenotyperメソッドよりも10〜20%遅くなりました。
一般に、選択されたモデルは問題の詳細とよく一致しており、ゲノム配列の分析に機械学習法を使用する一般的な傾向に対応しています。
この記事はバイオインフォマティクスワークショップの一部として書かれました。