これで一時停止が形成されたので、このトピックを終えて結果を示しました。
方法
(新しい読者に強制的に読まないように、前の記事から何かを繰り返します)
この記事の主な批判: 原核生物または「複数種起源」の進化的分類学に関する興味深い結果は 、「 1つの遺伝子を尺度と見なすことはできない 」という主張にありました 。 私はこれに完全に同意し、この実験はこれを完全に修正します。
いくつかの数字。 現在、NCBIには約2,000の細菌ゲノム(3,723遺伝子座)があります。 実験の準備として、この方法でラベル付けされたすべてのtRNAを選択しました。 彼らは4万以上のユニークなバリエーションであることが判明しました。 しかし、残念ながら、それらの間には多くの間違いがあります(約50%、これについて詳しく議論した以前の記事を参照)。
しかし、完全なエラー修正の段階はスキップできると思いました。 どうやってやるの? これらのtRNAを、長さおよび配列の最後のCCAの終わりの存在によってソートしました。 CCA配列はどのtRNAにも必要であり、長さは74から96ヌクレオチドであると言わなければなりません。
NCBIには、1ヌクレオチドからのtRNA、または1300を超えるまでの多くの奇跡があります:)(笑わずには言えません)。 したがって、最大70で100を超える長さのシーケンスと、CCAで終わらないシーケンスを削除しました。
それらは約20,000個あり、これらはNCBIからのエラーを含まない最も可能性の高いtRNAです。 tRNAの残りの半分を使用すると、後で把握できます。
実際、計画された実験では、この特定の配列が長さ70〜100ヌクレオチドを含むかどうかに違いはありません。 なんで? 2000個の細菌のゲノムを再確認するつもりなので、本当にそのような配列がありますか?エラーは除外されます。 そして、tRNAは実際には2番目のものかどうかです。 主なことは、DNAの重要な生物が異なる生物で一致することです。 ゲノム内の70〜100の配列長の一致は偶然とはほど遠いものです。
したがって、私は今何をしています。 私はこれらの20,000個のtRNAを取り、それらの中にどのバクテリアが存在するかを見つけます。 配列が1つの生物にのみ存在する場合、これは興味深いことではありません。 そして、おそらくこれは誤ったシーケンスです。 したがって、かなりの割合のエラーが排除されます。
シーケンスが複数の生物にある場合、これは2つの生物間の1つの関連付け(接続)です。
結果
最初の記事は重要な結論を下しました
複数種の起源は進化の状況を大きく混乱させますが、それについて行うことは何もありません-それは種分化の複雑さです。
したがって、適切な説明のために、系統樹的に系統樹は必要ありません。 最低でも、2つの親(平均化のため)、および一般的な場合はグラフと性器樹について話すことができます。
また、Graphvizを使用してグラフを表示することをお勧めしました。 しかし、グラフ内のリンクの数が1000を超えると、Graphvizがフリーズします。そして、6172個のリンクの合計グラフを取得しました。 したがって、ここでは明確にするために小さな断片のみを示します。 そして、私はほぼ1000リンクのグラフへのリンクを与えます。
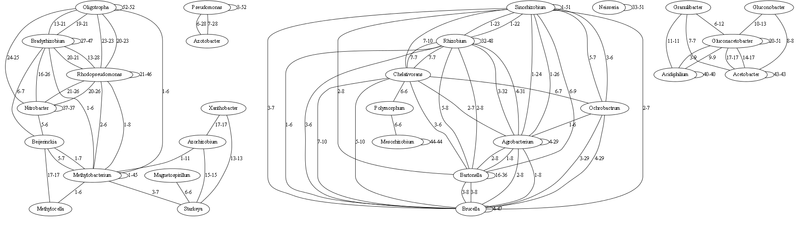
これが最強の結合を含むグラフです(5つの同一のtRNAへのリンクを含む)
各リンクは、一致する(100%同一の)tRNA遺伝子の最小最大数によって特徴付けられます。 属とそれ自体の関係は、この属内の同一のtRNAの数(つまり、種の違い)を意味します。
いくつかの結論
実際、このすべてを視覚的に処理する必要があるため、視覚的にこの多数をすべて受け入れることができます。 1000リンクのグラフには、誰とも接続されていない多くの属がありますが、最大5つの同一のtRNAでより弱いリンクを表示すると、遠い親relativeが表示されます。 (支援したい人がいる場合は、次のステップとしてこれを行うことを考えています-書き込み)。
実際、これに基づいて、現在の分類によると多くが一致しています。 同一のtRNAの数は、互いの属の範囲をよく示しており、同一のtRNAが少ないほど、古代の祖先が多くなります。 接続がほとんどないこれらの属は、最も古いものです(それらは現在シーケンス中であり、現在それらの個体群は別々の種によって表されているため)。 それらを分析することにより、初期進化のプロセスを非常に正確に構築できます。
更新しました。 グラフから双方向接続を削除しました(画像が詰まりました)。 リンクの総数は4551に減少しました。これにより、より大きなグラフを表示できました。
ここから画像をダウンロードできます(11.2 MB)。 以下は、最強の結合を含むグラフです(3つの同一のtRNAへのリンクを含む)
{kind=link}
次に、2つの巨大なドメイン間の接続(中間種)が表示されます(これらは画像で際立っており、おそらくベータおよびガンマプロテオバクテリアに対応しています)およびその他の詳細。 これが現在の分類にどの程度対応しているかを比較する必要がありますが、考えなければならないことがあります(詳細は、おそらく現在の科学的分類に該当しないものがあるようなものです)。
upd2 yEd Graph Editorを使用すると、完全なグラフが表示されます。 以下はミニ写真です。

接続が原因で画像がひどくなり、詳細が表示されないため、ファイルは下のyEd Graph Editor形式になります。少なくとも、拡大、移動、作成できます。 誰かが興味を持ち、より見やすいグラフを作成した場合-私は感謝します:)。
「原核生物の分類学(505属およびそれらの間の4548接続)」を数える