第二段階では、tRNAのフォールディングから始めました。 いくつかの問題がありました。 一方、これらの問題の解決に役立つ興味深いCRAアルゴリズムがあります。 複雑でわかりません。 ただし、主にLinux用の一部のソフトウェアに実装されています。 大きなファイとは何ですか。 一般的に、すべてが順番に。
PS数学を理解し、CRAアルゴリズムを理解するのに役立つ人を探しています。 一方、Gromacsを使用した人の助けが必要です。
TRNAフォールディング
モデル構造を実際の構造と比較できるように、既知のtRNA構造を取るように求められました。 今のところ、完全な結果は得られていません。 しかし同時に、誰かが他の有名なソフトウェアでシミュレーションを実行して結果を表示するのではないかと思います。 私の意見では、結果はないはずです。よく知られているソフトウェアでは、tRNAを適切にフォールディングできないものはないようです。
二次構造の取り扱いを開始します(図を参照)。これは、ヘリカル間非標準水素結合によって補完されます(これらは文献と、このtRNAの実際の構造を含む.pdbファイル内の結合測定に基づいています)。 私のアプローチでは、これらの水素結合と他のスタッキング相互作用結合(本質的に中間の折り畳み段階でのみ必要)を形成できる場合、実際の構造に非常に近いモデル構造が得られると主張しています。
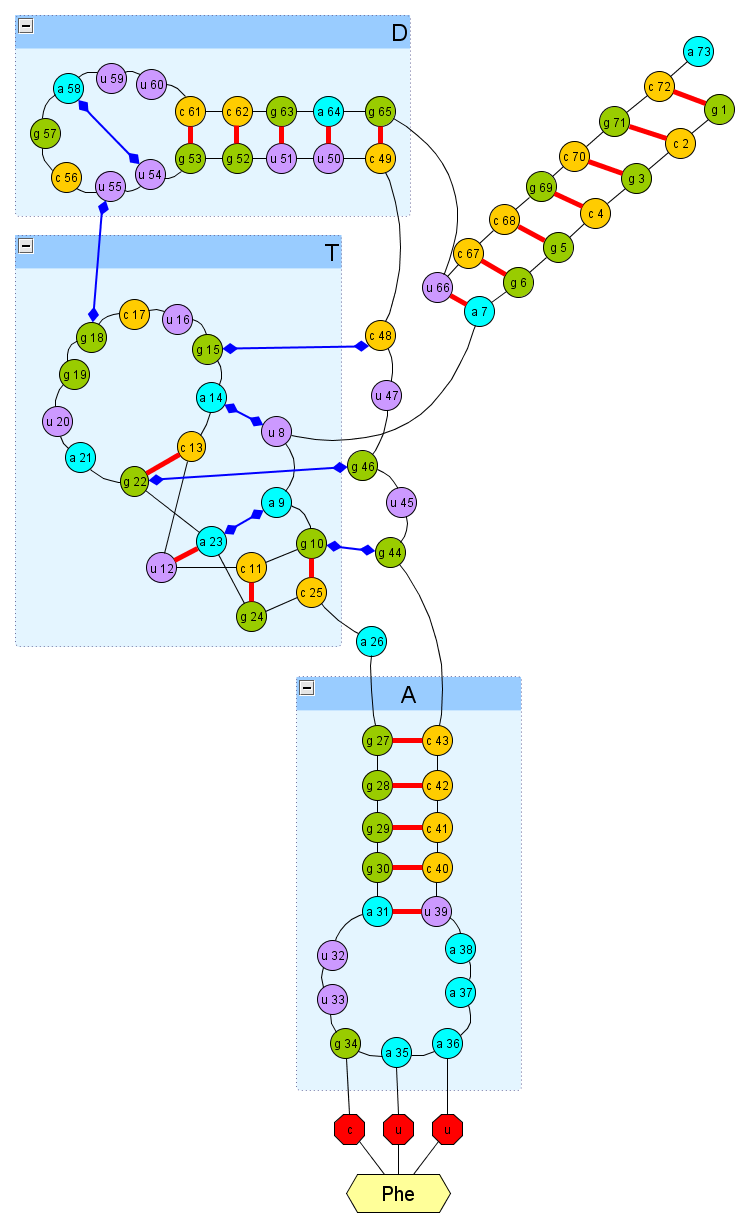
tRNAでは、すべてがリボザイムに関する前回の記事よりもはるかに複雑であることがわかります。 リボザイムで3つのヌクレオチド間で結合を形成するのが比較的困難だった場合、2つのヘリックスを正しく結合するには、t-ヘリックスが44-48ヌクレオチド、8-9で明確にラップされ、Dヘリックスに正しくドッキングされている必要があります。
詳細は説明しませんが、ここでの折り畳みシーケンスは単純ではなく、いわゆる 階層的な折り畳みモデル。これにより、最初にらせん(二次構造)が形成され、その後、より緊密に結合されます。 そうではありません。 スパイラルがすでに形成されている場合、ドッキングの可能性は事実上不可能になります。
現時点では、最後のスパイラル(1-7 + 66-72)を除くすべてのスパイラルが形成されているモデルを表示できます。 ただし、必要に応じて、すべての水素結合を作成するためのTヘリックスとDヘリックスのドッキングはまだ完全には行われていません。
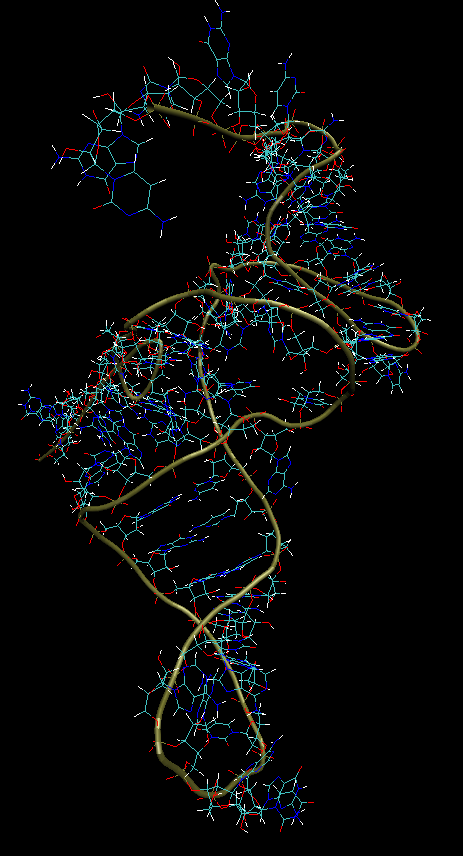
残りのヘリックスを正しくドッキングしてねじることができた場合、tRNAプロファイルは実際のプロファイルに対応していることがわかります。 そして、RMSDで測定すると、これは実質的に二次的な質問です。 しかし、それはこの分野の現在の科学者が比較される方法です。 このRNAの実際の.pdbモデルを研究しているときに、いくつかのエラーも発見しました。 したがって、生物学的方法によって得られるものの、理想的でもありません。 したがって、モデルを使用したRMSDによる測定は、慎重に信頼する必要があります。
CRAメソッド
彼はなぜですか?
もちろん、それをさらに数か月与えて、RNAInSpaceソフトウェアを使用して半自動で折り畳むことにより、TとDスパイラルのドッキングを達成することができます。折り畳みが間違っていることがわかる場所に介入し、折り畳みシーケンスと振動パラメーターを再生します。
ただし、自動化することはできません。 もちろん、 別のスパイラルに折り畳みます-今では自動化されていますが、スパイラルが結合されると、特定のヌクレオチド配列に対して別の完全にユニークな方法にスパイラルが合うはずです。 これらのらせんは互いにこすり合う必要があります。
そして、ここで私は長い間感じていた問題が発生しましたが、十分に明確に表現できませんでした。 以下に抜粋とリンクを示します。
基本的な問題の1つは、このタスクで「2人の教師を教える」という問題の説明に最初に触れることです。 また、「逆運動学」(ロボット工学の関節の計算に使用される)を使用する可能性についても議論しましたが、これはすべて非常に遅いです。 したがって、開発された方法は、RNAフォールディングの問題だけでなく、ロボット工学の分野の問題の解決策を改善するためにも適用できます。 ちなみに、折り畳みは、2つのヌクレオチドが接触する個々のケースで「車を駐車する」という問題を解決することに似ています。
一般に、それは驚くべきことですが、工学の分野でも、運動学に関連する他の分野でも、そのようなアルゴリズムは開発されていません。
私は、私が必要とする問題がどのように解決されるかを検索し、見つけることを試みました。 そのようなアルゴリズムがあり、長い間開発されてきましたが、最近実際に使用されています。 ロシア語では、いつものように:)近いものすらありません...これはやや不快です。
彼は具体的に何をしていますか?
このようなアルゴリズムは、調整された回転アルゴリズムと呼ばれます。 いわゆるについての記事に出会いました CRAアルゴリズムを改善するために作成されたCRISPアルゴリズム。 PHAISTOSで行われたCRISPとCRAの実装は、タンパク質構造シミュレーション用のマルコフ連鎖モンテカルロフレームワークです。 。
RNA(およびタンパク質)の主鎖は、結合で接続された原子で構成されています。 結合の長さは変更できませんが、原子のみがねじれます。 したがって、ある種の原子を回転させると、鎖全体がこの原子の後に強制的に回転します( 「my」リボザイムの折り畳みの軌跡上で、一方の端が固定され、もう一方の端が動いていることがわかります。タスクは両端を正しい位置に固定することです)。
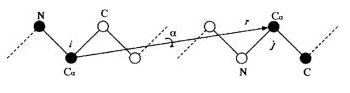
しかし、RNAを折り畳む場合、多くの場合、両側の原子を固定し、内側のギャップのみを回転させる必要があります。 これが私が「1つの根本的な問題」と呼んだものです。 これらのアルゴリズムにより、これを行うことができます。
これは2段階で行われます。 1つ目-アトムが欲しいときに回転します。 一緒にロックされている端の残りを回転させないと、チェーンが壊れます。 OK しかし、次のステップである次の原子の回転では、ギャップが完全になくなるように計算する必要があります。 私が理解しているように、ある種のエラーがあり、ギャップを閉じると原子の交差(結合)につながる可能性があるという事実に問題があります-したがって、異なる効率の異なるアルゴリズムがそれを行います。
彼はこれをどのようにしていますか?
しかし、これが問題です。 ここで私を助けてくれる数学者がいるかもしれません。式を見て、恐ろしくて吐き気を催したと思いました。 少なくとも誰かが助けるまで。 しかし、知識と数学者がいないこと、およびこのアルゴリズムの重要性を理解するために、既製の実装を調べに行きました。
実装の問題
最初の問題は、それらがLinuxで書かれていることです。 良いニュースは、それらがCで書かれていることです。 一般に、Linuxで書かれたコードを見ると、先史時代に戻っているという印象を克服することはできません。 Windows(つまり、おそらくマルチプラットフォーム)との互換性について話すことは完全に問題外です(そして、少なくとも3つの大きなプロジェクトを「見ました」)。
なぜ人々が過去30年生きたいのかさえ理解していません。 しかし、今はそれについてではありません。
第二に、コードの品質-Phaistos and Gromacs(これについては後ほど)-は単に悪いだけでなく、ひどいものです。 クラスやオブジェクトがない場合、または構造スタイルと絡み合っている場合は悪いです:)、すべてが絡み合うように書かれているのは恐怖です。
要するに、個別に実装されたメソッドを見つけるのは難しく、パッケージ全体を見て何かを理解する必要があります。 そして、これらは分子モデリングプロジェクト全体です。
PhaistosをWindowsにインストールしようとしました...不可能です。 ブーストライブラリを使用します 。数日間の苦労の末、私は明確な結論に達しましたが、ライブラリ自体はゴミであり(コードがひどくなるほどの量のテンプレートを使用しています)、MS Visual Studio上のWindowsではコンパイルされません。 忘れた
次に、CRAメソッドの別の実装に出会えて幸運でした。 しかし、健全なGromacsプロジェクトの下でも。 Windowsにインストールすることができ、実質的に判明します。 Gromacsを使用するコツを扱う必要があるのはここだけです。 たぶん彼と一緒に働いた人がいるのでしょうか?
そうすれば、プロセスが私にとってより速くなるかもしれません。 MS Visual StudioでCRAメソッドの実装を別のライブラリとして作成したいので、原子座標、固定原子、目的の回転を入力し、アルゴリズムが再びギャップを閉じます。