Prions, calcium, microbiota, food hormones and Alzheimer's disease
The fight against aging has several directions. One of them is age-related neurodegenerative pathologies, Alzheimer's and Parkinson’s diseases. Despite the active study of their molecular mechanisms, they have not yet learned to resist them. And this is becoming a big problem for older people who are most prone to neurodegeneration. Failures with therapy against neurodegeneration stimulate scientists to search for other key factors involved in the pathogenesis of neuropathologies. We will consider some of them with this review.
Alzheimer's disease (AD) is the most common form of dementia in the elderly with the clinical manifestations of progressive cognitive and functional impairment. Today, according to official figures, there are 47.5 million people with dementia in the world. 7.7 million new cases are registered annually. And it is expected that the number of people with dementia will increase to 75.6 million by 2030, and by 2050 - 135.5 million. Today, age is considered the main factor in increasing the risk of AD.
Pathology AD is usually characterized by extracellular accumulations of β-amyloid peptides (Aβ) in senile plaques and intracellular deposits of hyperphosphorylated tau protein, which forms neurofibrillary tangles. Genetically, apolipoprotein E alleles (APOE) (ε2, ε3 and ε4) carry various risks for the development of AD. Persons with the ε-4 allele have an increased risk compared with people with the more common ε3 allele, while the ε2 allele is associated with a reduced risk [1]. Not so long ago, it turned out that the pathogenesis of AD can have a long history and deposition of amyloid in the brain can precede clinical symptoms by 10–20 years [2]. There is evidence that AD is associated with chronic inflammation both in the central nervous system and in the periphery [3-5]. Despite a long study, treatment for AD does not yet exist. All attempts to create effective therapy have so far failed. Therefore, scientists are looking for new approaches to this disease.
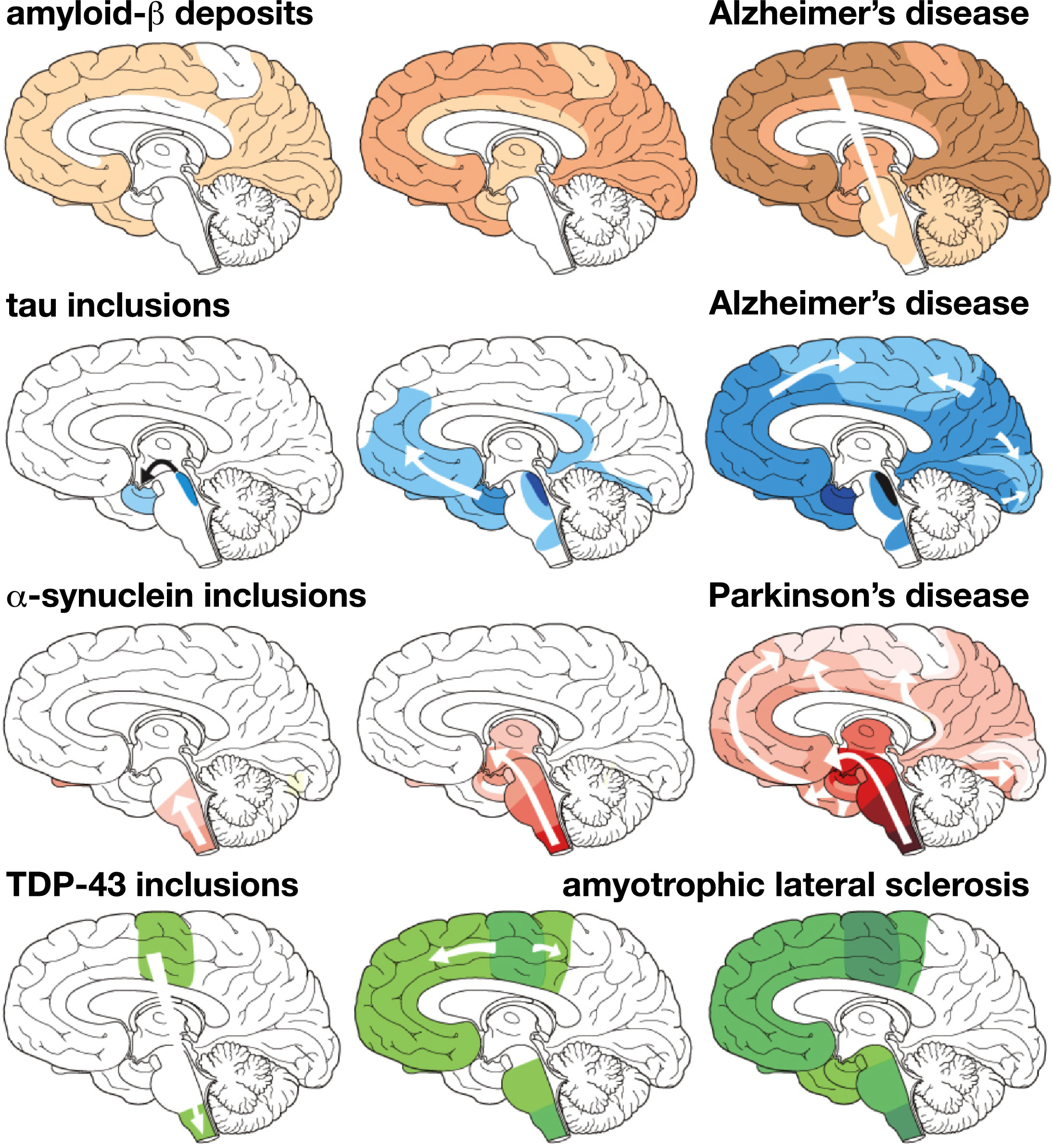
Fig. 1 The characteristic progression of specific protein lesions in neurodegenerative diseases, determined from post-mortem brain tests. Amyloid beta deposits in Alzheimer's disease; tau protein in Alzheimer's disease; α-synuclein in Parkinson's disease; and TDP-43 for amyotrophic lateral sclerosis. Three stages of progressive neurodegeneration, from left to right, with white arrows indicating the alleged spread of lesions [54].
So, in August this year, Nature Communications published an article by John W. Elrod et al., “Violation of mitochondrial outflow of calcium contributes to the progression of the disease in models of Alzheimer's disease.” In it, on the basis of the results obtained, the authors hypothesized the paramount importance of mitochondrial dysfunction and calcium overload in the development of Alzheimer's disease [6].
Next is a quote from the Temple University Health System release to this article.
“Sometimes, the more a person tries to solve a seemingly insignificant problem, the worse it gets. It turns out that the cells act similarly, and trying to compensate for what begins as a minor deficiency or dysfunction can become terrible.
In the case of Alzheimer's disease, the researchers showed that remodeling of mitochondrial calcium transport - which seems to be an attempt by cells to compensate for reduced energy production and metabolic dysfunction - may initially be useful, eventually becoming non-adaptive, contributing to a decrease in mitochondrial function and cognitive abilities .
A new study published in the journal Nature Communications is the first to link non-adaptive changes in calcium transport in mitochondria - energy-generating cells - to the progression of Alzheimer's disease.
Here is how the study leader, John W. Elrod, explains the essence of the discovery: “Amyloid deposition and tau pathology are considered the main causes of Alzheimer's disease and, as a result, they are the main direction of development of therapy. However, major clinical trials aimed at these pathways have failed everywhere. ”
“But so far no one has investigated the effect of altered calcium transport
to and from mitochondria on the progression of Alzheimer's disease. Our current study provides the missing link between these two hypotheses for the pathogenesis of Alzheimer's. ”
Calcium transport in mitochondria plays an important role in many cellular functions and requires the effective participation of multiple proteins. One of the key regulators of this process is a protein known as NCLX, which was previously discovered in the laboratory of J. Elrod as a participant in the regulation of calcium transport from heart cells. NCLX expression is also important for mitochondrial calcium transport in neurons.
In a new study, John W. Elrod and colleagues examined the role of mitochondrial calcium uptake by neurons in Alzheimer's disease. To do this, the team used a mouse model of Alzheimer's disease, in which animals possessed three gene mutations that cause age-related pathology comparable to the progression of Alzheimer's disease in humans.
As mice bearing three mutations age, the researchers observed a steady decrease in NCLX expression. This decrease was accompanied by a decrease in the expression of proteins that limit the accumulation of mitochondrial calcium, which leads to devastating calcium overload. The loss of NCLX was also associated with increased production of cell-damaging oxidizing agents.
To better understand the physiological significance of NCLX loss, scientists completely suppressed the expression of NCLX in the brain of Alzheimer's mice. Analysis of the brain tissue of these mice showed that a decrease in NCLX and subsequent loss of calcium outflow from mitochondria accelerate the accumulation of amyloid beta and tau pathology.
“Our results show that maladaptive remodeling of pathways to compensate for dysregulation of calcium, which may be designed to maintain energy production in cells, leads to neuronal dysfunction and Alzheimer's disease pathology. Moreover, our data indicate that amyloid beta accumulation and tau pathology actually lie below mitochondrial dysfunction in the progression of Alzheimer's disease, which opens up a new therapeutic direction. ” [7]
In addition to mitochondria and calcium, possibly a diet and intestinal microbiota also significantly affect the development of AD. Since 2014, researchers have shown the potential role of pathogenic microbes, including intestinal, in the development and progression of AD. As it is already known today, intestinal microbiota is an important component of human health. Intestinal microbes perform key functions for human health, including energy extraction, biosynthesis of vitamins, protection against excessive growth of pathogens, etc. Microbial colonization of the intestine occurs during childbirth, is very dynamic in infancy, and develops into an adult structure by about 3 years of age.
It has been established that changes in the composition of a complex microbial ecosystem are associated with the development of various gastrointestinal and metabolic diseases, including inflammatory bowel disease, obesity, insulin resistance, and diabetes [8]. Recently, much attention has been paid to the influence of the intestinal microbiota on the function of the central nervous system, which is often called the axis of the intestine - the brain. Changes in gut microbiome are associated with neurological conditions, including autism spectrum disorders, multiple sclerosis, and Parkinson's disease [9-11].
Studies in animal models of AD have shown that changes in the intestinal microbiota can affect the rate of deposition of amyloid [12,13]. In people with AD, scientists observed a decrease in the microbial diversity of the intestinal microbiome, in contrast to a control group consisting of people of age and gender. Differences between bacterial types were detected, including a decrease in Firmicutes and Actinobacteria (including Bifidobacteriaceae) and an increase in Bacteroidetes in the microbiome of people with AD [14].
Together with other factors, diet may play a role in AD pathology. An increased risk of developing dementia is associated with a diet high in saturated fats and simple carbohydrates, what is called a “western” diet. Conversely, diets with a high content of mono- and polyunsaturated fats, vegetables, fruits, and low-fat proteins are associated with a reduced risk of AD [15-17].
In 2013, UNESCO included the Mediterranean diet in the “Representative List of the Intangible Cultural Heritage of Humanity” [18]. The Mediterranean-style diet is a balanced diet characterized by high intake of fiber, olive oil, fruits, nuts, vegetables and cereals; moderate consumption of fish and poultry; low consumption of dairy products, red meat, meat products and sweets. And also the consumption of wine in moderation [19]. The Mediterranean diet has been found to be associated with a decrease in the incidence of certain chronic diseases such as obesity, type 2 diabetes, cancer, cardiovascular and neurodegenerative diseases, including Alzheimer's disease [17, 20, 21].
Although the underlying mechanisms remain unclear, recent evidence suggests that modulation of the intestinal microbiome and microbial metabolites is one of the possible factors mediating the health effects of the Mediterranean diet [20, 22].
In people with mild Congestive Disorders, scientists have found that certain bacteria are associated with markers of Alzheimer's disease in the cerebrospinal fluid. A modified Mediterranean ketogenic diet (MMKD) altered intestinal microbiomes and decreased levels of one of the major markers of AD, Aß42. MMKD was reduced in carbohydrates, with the main part of the diet being fats and proteins derived from olive oil and fish. Several mechanisms of the action of the ketogenic diet, including a decrease in neuronal hyper-excitability, an increase in mitochondrial metabolism, a decrease in oxidative stress, and inhibition of mTOR, can affect pathological processes in AD [23].
After 6 weeks of MMKD diet, participants with mild cognitive impairment, which often precede AD, showed a decrease in lactate and an increase in butyrate, as well as positive changes in the intestinal microbiome and a decrease in cerebrospinal fluid Aß42 [24].
In confirmation of the important role of intestinal microbiota in the pathogenesis of AD, three studies on transplantation of fecal microbiota were published immediately this year. Normal gut microbiota from healthy mice was transplanted into transgenic mice with the AD model. In all cases, a marked improvement in the condition of animals with AD occurred: “Our results showed that the use of FMT (fecal microbiota transplantation) can improve cognitive deficit and reduce deposition of amyloid (Aβ) in the brain of transgenic (Tg) APPswe / PS1dE9 mice. These improvements were accompanied by a decrease in tau protein phosphorylation and Aβ40 and Aβ42 levels. We observed an increase in synaptic plasticity in Tg mice, while PSD-95 protein expression and synapsin I expression increased after FMT. We also observed a decrease in COX-2 and CD11b levels in Tg mice after FMT. We also found that the use of FMT caused changes in the intestinal microbiota and SCFAs (short chain fatty acids). Therefore, FMT may be a potential therapeutic strategy for AD ”[42-44].
Also this year came another important piece of news on Alzheimer's. The first prospective study was published showing an increased risk of AD after a blood transfusion. The study group consisted of 63,813 patients who underwent a blood transfusion, and the same number of people in the control group. The follow-up period was 10 years. The results showed that people who have undergone a blood transfusion have a 1.73-fold higher risk of dementia and a 1.37-fold higher risk of Alzheimer's than those who did not. Patients who received a washed red blood cell transfusion had a 2.37-fold higher risk of developing dementia compared to those who did not [45].
Although the authors are cautious in their conclusions, one of the possible reasons for this important discovery may be the prion nature of Alzheimer's disease. And this is worth telling in more detail.
One of the first breakthrough works on this topic was published back in 2015 in the journal Nature. Sebastian Brandner and colleagues examined post-mortem brain samples of people who died from Creutzfeldt-Jakob disease, or "mad cow disease." This disease has a prion nature. People whose brains were studied by scientists became infected as a result of injections of growth hormone prion-infected. They had an average age of 36-51 years, and besides prions, their brain tissue contained accumulations of amyloid beta. All these people did not have a genetic predisposition to Alzheimer's disease. In addition, scientists examined 116 patients with other prion diseases of the same age and over 10 years of age. None of the examined showed amyloid beta accumulations in the brain. This allowed scientists to suggest that amyloid beta appeared in the brain of people with Creutzfeldt-Jakob disease for the same reason as prions - as a result of infection with growth hormones by infected injections [46].
In 2018, two more works of this team of scientists came out. Nature published their article in which they reported the results of a study of growth hormone samples obtained from the human pituitary gland that caused the development of prion disease. “Given the importance of our hypothesis for public health, we have begun the identification and biochemical analysis of archival vials of c-hGH (growth hormone). Here we show that certain batches of c-hGH that have been used by patients with Creutzfeldt-Jakob pathology and Aβ clusters have significant levels of Aβ40, Aβ 42, and tau proteins. And this material can cause the formation of Aβ plaques and cerebral amyloid angiopathy in mice expressing the human amyloid precursor protein. These results confirm the presence of Aβ in archived c-hGH vials and are consistent with the alleged iatrogenic (ie, mediated through medical procedures) transmission of Aβ pathology to humans ”, the study authors conclude [47].
In the second work of the same year, S. Brandner et al. provided data showing the possibility of infection with beta-amyloids during neurosurgical operations. “Here we present four patients who underwent neurosurgical procedures in childhood or adolescence and received about three decades later intracerebral hemorrhage caused by severe cerebral amyloid angiopathy (CAA). None of these patients had pathogenic mutations associated with the early development of Aβ pathology. In addition, we identified four patients with a history of neurosurgical intervention and subsequent development of CAA in the literature ”[48].
A year before, a sensational study by Chinese scientists came to be a sensation. Blood flow was combined in healthy and transgenic mice with AD, resulting in amyloid beta accumulations in wild normal animals in the brain. In fact, amyloid infection occurred through the blood from a sick animal to a healthy one. “In this study, using a parabiosis model between APsswe / PS1dE9 AP transgenic mice and their wild counterparts, we found that human Aβ, derived from transgenic AD model mice, entered the blood circulation and accumulated in the brain of wild-type mice, and cerebral amyloid angiopathy formed and Aβ plaques after a 12-month period of parabiosis. Pathologies of type AD associated with the accumulation of Aβ, including tau hyperphosphorylation, neurodegeneration, inflammation and microhemorrhages, have been found in the brains of wild-type parabiotic mice. As far as we know, our study is the first to reveal that Aβ originating from the blood can penetrate the brain, form Aβ-related pathologies and cause functional neuron deficiency ”[49].
The weight of this hypothesis about the possibility of infection and the further spread of beta-amyloid according to the prion principle is given by the fact that it is supported by the discoverer of prions, Nobel laureate Stanley Pruziner. In 2019, the work of S. Pruziner and his colleagues appeared in Science Translational Medicine, in which they provided new evidence for this hypothesis [50].
What attracts attention: the second and third most common neuropathology after AD, Parkinson's disease and amyotrophic lateral sclerosis, are also possibly spread throughout the body in a prion-like manner. And there are a number of studies about this [51, 52]. Thus, clusters of alpha-synuclein, the main participant in Parkinson's disease, as described in a recent work in 2019, first appear in the intestine and then move through other tissues like prions. Moreover, these clusters in the gastrointestinal tract can be detected, according to scientists, 20 years before the onset of clinical symptoms of Parkinson's disease. “ In this new study, we found out exactly how the disease can spread from the intestines of people. We probably will not be able to develop effective treatments that will stop the disease without knowing where it starts and how it spreads - so this is an important step in our study of pathology. Parkinson's disease is a complex disease that we are still trying to understand. However, thanks to this study and a similar study in the USA, which recently led to the same result using mice, it is suggested that the disease begins in the intestines of patients, ”says one of the authors of the work, P. Borghammer [53].
Interesting data on similar "related" mechanisms of the pathogenesis of Alzheimer's and Parkinson's diseases were obtained by Z. Zhang et al. They showed that the same enzyme, asparagine endopeptidase (AEP), cleaves both the tau protein in AD and alpha-synuclein in PD, which leads to their aggregation and neurotoxicity [54].
It is quite obvious that the prion-like distribution of the three main neurodegenerative pathologies can hardly be an accident. Although the possibility of infection from the outside is shown today only for Alzheimer's disease. In 2019, American scientists, J. Weickenmeier et al., Were able to bring the theoretical base to the prion hypothesis for these three neuropathologies. They created a physical model for them that explains the prion-like features of neurodegeneration in Alzheimer's, Parkinson’s and amyotrophic lateral sclerosis: “ Our results show that improperly folded proteins in various neurodegenerative diseases grow and spread in accordance with the universal law, which follows the basic physical principles of nonlinear reaction and anisotropic diffusion. Our results confirm the concept of the general fundamental principle of the pathogenesis of a wide range of neurodegenerative disorders - prion paradigm . " Fans of complex formulas and equations will be able to enjoy reading this article [55].
Well, in addition to prions, we will tell you about one more discovered interesting feature of the brain of people with AD. As shown by British scientists, Shelley J. Allen et al., The brain tissue of patients with AD contains an abnormally large number of microbes skewed towards P. acnes . " P. acnes is a commensal gram-positive component of the microflora of the human skin and mouth, preferring anaerobic growth conditions, and it is becoming increasingly apparent that it is a significant opportunistic microorganism. This is most often associated with postoperative lesions and implanted prostheses, as well as chronic diseases such as inflammation of the lumbar, endocarditis, sarcoidosis, and intracranial lesions. Recently, the presence of proteobacteria and actinobacteria (containing Propionibacteriaceae) in mo n, affected both normal and multiple sclerosis; thus, the normal brain has germs, consisting mainly of Proteobacteria and Actinobacteria Our data suggest that Actinobacteria (P. acnes) increases in the brain in AD brain damage proteobacteria ability.. P. acnes nonspecifically stimulate the innate immune system is well-documented "- the authors of [56] write.
Also in recent work, scientists have found that the hunger hormone ghrelin may be involved in the development of Alzheimer's disease. Ghrelin receptors have recently been discovered in the hippocampus. The hippocampus is an area of the brain that is important for learning, memory and emotions. In AD, this is one of the first areas where cell death and damage due to the formation of amyloid beta accumulations occurs. In a healthy hippocampus, ghrelin binds to its receptor, GHSR1α. This then mediates the activation of the dopamine receptor D1 (DRD1). Modulation of DRD1 by GHSR1α is critical for the function of the hippocampal synapses and synaptic reorganization via the Gαq-Ca 2+ signaling pathway, which is central to memory formation.
The role of GHSR1α in the synaptic function of the hippocampus has allowed scientists to suggest that receptor dysfunction may contribute to the synaptic deficit in the hippocampus observed in AD.
Researchers at the University of Dallas studied this issue using brain samples from people suffering from AD and an animal model from AD. The hypothesis of scientists was that dissociation (i.e. separation) between ghrelin and dopamine receptors may be the very factor that affects cognitive ability in patients with Alzheimer's disease.
Scientists have been able to find that amyloid β inhibits the activation of GHSR1α. Which in turn disrupts the GHSR1α-mediated activation of DRD1 in the hippocampus of people with AD. In an animal model, researchers injected two mice with AD activating the ghrelin and dopamine receptors in the hippocampus, MK0677 and SKF8129. This combination prevented inhibition of the GHSRr1α receptor by β-amyloid, mitigating synaptic damage to the hippocampus and improving learning and memory of rodents [25].
Characteristically, ghrelin is not the first “food" hormone that can be associated with cognitive impairment and Alzheimer's. Prior to this, they discovered the effect of insulin with resistance to it, even calling Alzheimer's type 3 diabetes.
As the available data show, AD can be a slowly progressive metabolic disease of the brain. And numerous studies demonstrate a complex relationship between metabolic syndrome (MetS) and AD. People with diabetes and obesity have a higher risk of developing AD. At the same time, patients with AD often develop hyperglycemia and insulin resistance (IR). IR as a violation of insulin signaling is a common characteristic of both MetS and AD. And, according to scientists, it is a key link between the two diseases. Insulin signaling regulates Aβ and tau levels, and Aβ has a negative effect on insulin signaling. Therefore, insulin signaling dysfunction can enhance the pathology of Aβ and tau, and increased production of Aβ can further exacerbate IR.The accumulated data also indicate that AD is closely related to dysfunction of both insulin signaling and glucose metabolism in the brain, which prompts some researchers to attribute AD to type 3 diabetes or to an insulin-resistant state of the brain [26, 27].
Insulin is secreted by the beta cells of the pancreas and enters the central nervous system, crossing the blood-brain barrier in a controlled and saturated manner. The synthesis of insulin in the brain itself is still a matter of discussion [28]. Insulin receptors (InsRs) are widely expressed in the brain, including the olfactory bulb, cerebral cortex, hippocampus, hypothalamus, and amygdala [29]. InsRs are more concentrated in neurons compared to glial cells [30].
The transmission of insulin signals to the brain plays an important role in regulating food intake, body weight, reproduction, learning and memory. Intranasal administration of insulin improves working memory in both human and animal studies [31]. In addition, the levels of InsR mRNA and protein increase in the CA1 region of the hippocampus during the formation of short-term memory [32]. This suggests that the sensitivity of neurons to insulin may be increased during training.
Impaired insulin signaling makes neurons more vulnerable to metabolic stress, accelerating neuronal dysfunction. Defective insulin signaling is associated with decreased cognitive abilities and the development of dementia, including AD [33]. The deterioration of cognitive abilities in diabetes and AD is associated with a decrease in InsR expression and insulin level in cerebrospinal fluid (CSF) [34, 35]. Decreased insulin signaling, including altered kinase activity and IRS expression, with AD worsens with disease progression [36,37]. And increased phosphorylation of IRS-1, a key IR factor, occurs in the brain with AD [38]. Interestingly, the brain regions with the highest InsR density, such as the hippocampus and temporal lobe, are also the main targets of neurodegeneration in AD [39, 40]. Therefore, impaired insulin signaling,caused by IR can have a big impact on cognitive decline and the development of AD.
Several diabetes treatments that enhance insulin signaling are being tested for therapeutic potential in AD and dementia. Although the results of clinical trials of TZD have been disappointing, intranasal insulin and GLP-1 analogues are still actively used as a potential treatment for AD and have shown some promising results. Intranasal insulin, however, is only effective in the early stages of AD. In addition, exenatide and liraglutide are still in the early stages of therapeutic development, and large clinical trials are currently underway [41].
But that is not all.It turns out that in addition to ghrelin and insulin, two more food hormones, among adipokines (adipose tissue hormones), leptin and adiponectin can participate in Alzheimer's.
There are recent studies from this year:
Obesity as a risk factor for Alzheimer's disease: the effects of leptin and glutamate.
The role of leptin and adiponectin in the cognitive decline associated with obesity and Alzheimer's disease.
Earlier work, 2016 and 2018:
Leptin dysfunction and Alzheimer's disease: data from cell studies, animal and human studies.
Hepocampus leptin regulation and its role in Alzheimer's disease.
There is increasing evidence that leptin has cognitively stimulating properties, as it is involved in the signaling pathways underlying hippocampal learning and memory. However, a significant decrease in the ability of leptin to regulate the synaptic function of the hippocampus occurs with age, and dysfunctions in the leptin system are associated with an increased risk of developing Alzheimer's disease.
www.ncbi.nlm.nih.gov/pubmed/28987937
Another hormone associated with adipose tissue and inflammation, resistin, plays an important role in inflammation of the hypothalamus and hormonal regulation: The
molecular mechanisms underlying obesity-induced inflammation of the hypothalamus and insulin resistance: a key role of the resistin / TLR4 pathway
www.ncbi.nlm.nih.gov/pmc/articles/PMC6418006
In the end, what happens? A number of “food hormones” (ghrelin, insulin, resistin, leptin, adiponectin), the regulation of which is disturbed during aging, can one way or another participate in neurodegenerative processes.
Therefore, it is not at all accidental that antidiabetic drugs, metformin, and others undergo preclinical studies with a view to counteracting age-related neurodegeneration.
Antidiabetic drugs for Alzheimer's disease: mechanisms of action and future prospects
Well, at the very end we’ll briefly say how a violation of sleep regimen and quality can be associated with the development of AD. In 2018, N. Volkow et al. Published in PNAS, in which they showed that even one sleepless night can increase the accumulation of amyloid beta in the brain [57].
And in 2019, scientists from the University of Washington published a study showing that a decrease in deep sleep is associated with early signs of Alzheimer's disease [58].
Summarize.The most common age-related neurodegenerative pathologies show the extremely complex nature of their pathogenesis. To understand which and put into a single picture, we are not yet able to. There are no effective medicines against them yet and it is not even clear on what basis they should be created. Therefore, what a person can do now is to lead a healthy lifestyle and support, to the extent possible, work and research directed against aging in general and age-related neurodegeneration in particular.
The review was prepared by: M. Batin, A. Rzheshevsky.
Alzheimer's disease (AD) is the most common form of dementia in the elderly with the clinical manifestations of progressive cognitive and functional impairment. Today, according to official figures, there are 47.5 million people with dementia in the world. 7.7 million new cases are registered annually. And it is expected that the number of people with dementia will increase to 75.6 million by 2030, and by 2050 - 135.5 million. Today, age is considered the main factor in increasing the risk of AD.
Pathology AD is usually characterized by extracellular accumulations of β-amyloid peptides (Aβ) in senile plaques and intracellular deposits of hyperphosphorylated tau protein, which forms neurofibrillary tangles. Genetically, apolipoprotein E alleles (APOE) (ε2, ε3 and ε4) carry various risks for the development of AD. Persons with the ε-4 allele have an increased risk compared with people with the more common ε3 allele, while the ε2 allele is associated with a reduced risk [1]. Not so long ago, it turned out that the pathogenesis of AD can have a long history and deposition of amyloid in the brain can precede clinical symptoms by 10–20 years [2]. There is evidence that AD is associated with chronic inflammation both in the central nervous system and in the periphery [3-5]. Despite a long study, treatment for AD does not yet exist. All attempts to create effective therapy have so far failed. Therefore, scientists are looking for new approaches to this disease.
Fig. 1 The characteristic progression of specific protein lesions in neurodegenerative diseases, determined from post-mortem brain tests. Amyloid beta deposits in Alzheimer's disease; tau protein in Alzheimer's disease; α-synuclein in Parkinson's disease; and TDP-43 for amyotrophic lateral sclerosis. Three stages of progressive neurodegeneration, from left to right, with white arrows indicating the alleged spread of lesions [54].
Calcium and mitochondrial dysfunction
So, in August this year, Nature Communications published an article by John W. Elrod et al., “Violation of mitochondrial outflow of calcium contributes to the progression of the disease in models of Alzheimer's disease.” In it, on the basis of the results obtained, the authors hypothesized the paramount importance of mitochondrial dysfunction and calcium overload in the development of Alzheimer's disease [6].
Next is a quote from the Temple University Health System release to this article.
“Sometimes, the more a person tries to solve a seemingly insignificant problem, the worse it gets. It turns out that the cells act similarly, and trying to compensate for what begins as a minor deficiency or dysfunction can become terrible.
In the case of Alzheimer's disease, the researchers showed that remodeling of mitochondrial calcium transport - which seems to be an attempt by cells to compensate for reduced energy production and metabolic dysfunction - may initially be useful, eventually becoming non-adaptive, contributing to a decrease in mitochondrial function and cognitive abilities .
A new study published in the journal Nature Communications is the first to link non-adaptive changes in calcium transport in mitochondria - energy-generating cells - to the progression of Alzheimer's disease.
Here is how the study leader, John W. Elrod, explains the essence of the discovery: “Amyloid deposition and tau pathology are considered the main causes of Alzheimer's disease and, as a result, they are the main direction of development of therapy. However, major clinical trials aimed at these pathways have failed everywhere. ”
“But so far no one has investigated the effect of altered calcium transport
to and from mitochondria on the progression of Alzheimer's disease. Our current study provides the missing link between these two hypotheses for the pathogenesis of Alzheimer's. ”
Calcium transport in mitochondria plays an important role in many cellular functions and requires the effective participation of multiple proteins. One of the key regulators of this process is a protein known as NCLX, which was previously discovered in the laboratory of J. Elrod as a participant in the regulation of calcium transport from heart cells. NCLX expression is also important for mitochondrial calcium transport in neurons.
In a new study, John W. Elrod and colleagues examined the role of mitochondrial calcium uptake by neurons in Alzheimer's disease. To do this, the team used a mouse model of Alzheimer's disease, in which animals possessed three gene mutations that cause age-related pathology comparable to the progression of Alzheimer's disease in humans.
As mice bearing three mutations age, the researchers observed a steady decrease in NCLX expression. This decrease was accompanied by a decrease in the expression of proteins that limit the accumulation of mitochondrial calcium, which leads to devastating calcium overload. The loss of NCLX was also associated with increased production of cell-damaging oxidizing agents.
To better understand the physiological significance of NCLX loss, scientists completely suppressed the expression of NCLX in the brain of Alzheimer's mice. Analysis of the brain tissue of these mice showed that a decrease in NCLX and subsequent loss of calcium outflow from mitochondria accelerate the accumulation of amyloid beta and tau pathology.
“Our results show that maladaptive remodeling of pathways to compensate for dysregulation of calcium, which may be designed to maintain energy production in cells, leads to neuronal dysfunction and Alzheimer's disease pathology. Moreover, our data indicate that amyloid beta accumulation and tau pathology actually lie below mitochondrial dysfunction in the progression of Alzheimer's disease, which opens up a new therapeutic direction. ” [7]
Intestinal microbiota and diet
In addition to mitochondria and calcium, possibly a diet and intestinal microbiota also significantly affect the development of AD. Since 2014, researchers have shown the potential role of pathogenic microbes, including intestinal, in the development and progression of AD. As it is already known today, intestinal microbiota is an important component of human health. Intestinal microbes perform key functions for human health, including energy extraction, biosynthesis of vitamins, protection against excessive growth of pathogens, etc. Microbial colonization of the intestine occurs during childbirth, is very dynamic in infancy, and develops into an adult structure by about 3 years of age.
It has been established that changes in the composition of a complex microbial ecosystem are associated with the development of various gastrointestinal and metabolic diseases, including inflammatory bowel disease, obesity, insulin resistance, and diabetes [8]. Recently, much attention has been paid to the influence of the intestinal microbiota on the function of the central nervous system, which is often called the axis of the intestine - the brain. Changes in gut microbiome are associated with neurological conditions, including autism spectrum disorders, multiple sclerosis, and Parkinson's disease [9-11].
Studies in animal models of AD have shown that changes in the intestinal microbiota can affect the rate of deposition of amyloid [12,13]. In people with AD, scientists observed a decrease in the microbial diversity of the intestinal microbiome, in contrast to a control group consisting of people of age and gender. Differences between bacterial types were detected, including a decrease in Firmicutes and Actinobacteria (including Bifidobacteriaceae) and an increase in Bacteroidetes in the microbiome of people with AD [14].
Together with other factors, diet may play a role in AD pathology. An increased risk of developing dementia is associated with a diet high in saturated fats and simple carbohydrates, what is called a “western” diet. Conversely, diets with a high content of mono- and polyunsaturated fats, vegetables, fruits, and low-fat proteins are associated with a reduced risk of AD [15-17].
In 2013, UNESCO included the Mediterranean diet in the “Representative List of the Intangible Cultural Heritage of Humanity” [18]. The Mediterranean-style diet is a balanced diet characterized by high intake of fiber, olive oil, fruits, nuts, vegetables and cereals; moderate consumption of fish and poultry; low consumption of dairy products, red meat, meat products and sweets. And also the consumption of wine in moderation [19]. The Mediterranean diet has been found to be associated with a decrease in the incidence of certain chronic diseases such as obesity, type 2 diabetes, cancer, cardiovascular and neurodegenerative diseases, including Alzheimer's disease [17, 20, 21].
Although the underlying mechanisms remain unclear, recent evidence suggests that modulation of the intestinal microbiome and microbial metabolites is one of the possible factors mediating the health effects of the Mediterranean diet [20, 22].
In people with mild Congestive Disorders, scientists have found that certain bacteria are associated with markers of Alzheimer's disease in the cerebrospinal fluid. A modified Mediterranean ketogenic diet (MMKD) altered intestinal microbiomes and decreased levels of one of the major markers of AD, Aß42. MMKD was reduced in carbohydrates, with the main part of the diet being fats and proteins derived from olive oil and fish. Several mechanisms of the action of the ketogenic diet, including a decrease in neuronal hyper-excitability, an increase in mitochondrial metabolism, a decrease in oxidative stress, and inhibition of mTOR, can affect pathological processes in AD [23].
After 6 weeks of MMKD diet, participants with mild cognitive impairment, which often precede AD, showed a decrease in lactate and an increase in butyrate, as well as positive changes in the intestinal microbiome and a decrease in cerebrospinal fluid Aß42 [24].
In confirmation of the important role of intestinal microbiota in the pathogenesis of AD, three studies on transplantation of fecal microbiota were published immediately this year. Normal gut microbiota from healthy mice was transplanted into transgenic mice with the AD model. In all cases, a marked improvement in the condition of animals with AD occurred: “Our results showed that the use of FMT (fecal microbiota transplantation) can improve cognitive deficit and reduce deposition of amyloid (Aβ) in the brain of transgenic (Tg) APPswe / PS1dE9 mice. These improvements were accompanied by a decrease in tau protein phosphorylation and Aβ40 and Aβ42 levels. We observed an increase in synaptic plasticity in Tg mice, while PSD-95 protein expression and synapsin I expression increased after FMT. We also observed a decrease in COX-2 and CD11b levels in Tg mice after FMT. We also found that the use of FMT caused changes in the intestinal microbiota and SCFAs (short chain fatty acids). Therefore, FMT may be a potential therapeutic strategy for AD ”[42-44].
Prion nature of neuropathologies
Also this year came another important piece of news on Alzheimer's. The first prospective study was published showing an increased risk of AD after a blood transfusion. The study group consisted of 63,813 patients who underwent a blood transfusion, and the same number of people in the control group. The follow-up period was 10 years. The results showed that people who have undergone a blood transfusion have a 1.73-fold higher risk of dementia and a 1.37-fold higher risk of Alzheimer's than those who did not. Patients who received a washed red blood cell transfusion had a 2.37-fold higher risk of developing dementia compared to those who did not [45].
Although the authors are cautious in their conclusions, one of the possible reasons for this important discovery may be the prion nature of Alzheimer's disease. And this is worth telling in more detail.
One of the first breakthrough works on this topic was published back in 2015 in the journal Nature. Sebastian Brandner and colleagues examined post-mortem brain samples of people who died from Creutzfeldt-Jakob disease, or "mad cow disease." This disease has a prion nature. People whose brains were studied by scientists became infected as a result of injections of growth hormone prion-infected. They had an average age of 36-51 years, and besides prions, their brain tissue contained accumulations of amyloid beta. All these people did not have a genetic predisposition to Alzheimer's disease. In addition, scientists examined 116 patients with other prion diseases of the same age and over 10 years of age. None of the examined showed amyloid beta accumulations in the brain. This allowed scientists to suggest that amyloid beta appeared in the brain of people with Creutzfeldt-Jakob disease for the same reason as prions - as a result of infection with growth hormones by infected injections [46].
In 2018, two more works of this team of scientists came out. Nature published their article in which they reported the results of a study of growth hormone samples obtained from the human pituitary gland that caused the development of prion disease. “Given the importance of our hypothesis for public health, we have begun the identification and biochemical analysis of archival vials of c-hGH (growth hormone). Here we show that certain batches of c-hGH that have been used by patients with Creutzfeldt-Jakob pathology and Aβ clusters have significant levels of Aβ40, Aβ 42, and tau proteins. And this material can cause the formation of Aβ plaques and cerebral amyloid angiopathy in mice expressing the human amyloid precursor protein. These results confirm the presence of Aβ in archived c-hGH vials and are consistent with the alleged iatrogenic (ie, mediated through medical procedures) transmission of Aβ pathology to humans ”, the study authors conclude [47].
In the second work of the same year, S. Brandner et al. provided data showing the possibility of infection with beta-amyloids during neurosurgical operations. “Here we present four patients who underwent neurosurgical procedures in childhood or adolescence and received about three decades later intracerebral hemorrhage caused by severe cerebral amyloid angiopathy (CAA). None of these patients had pathogenic mutations associated with the early development of Aβ pathology. In addition, we identified four patients with a history of neurosurgical intervention and subsequent development of CAA in the literature ”[48].
A year before, a sensational study by Chinese scientists came to be a sensation. Blood flow was combined in healthy and transgenic mice with AD, resulting in amyloid beta accumulations in wild normal animals in the brain. In fact, amyloid infection occurred through the blood from a sick animal to a healthy one. “In this study, using a parabiosis model between APsswe / PS1dE9 AP transgenic mice and their wild counterparts, we found that human Aβ, derived from transgenic AD model mice, entered the blood circulation and accumulated in the brain of wild-type mice, and cerebral amyloid angiopathy formed and Aβ plaques after a 12-month period of parabiosis. Pathologies of type AD associated with the accumulation of Aβ, including tau hyperphosphorylation, neurodegeneration, inflammation and microhemorrhages, have been found in the brains of wild-type parabiotic mice. As far as we know, our study is the first to reveal that Aβ originating from the blood can penetrate the brain, form Aβ-related pathologies and cause functional neuron deficiency ”[49].
The weight of this hypothesis about the possibility of infection and the further spread of beta-amyloid according to the prion principle is given by the fact that it is supported by the discoverer of prions, Nobel laureate Stanley Pruziner. In 2019, the work of S. Pruziner and his colleagues appeared in Science Translational Medicine, in which they provided new evidence for this hypothesis [50].
What attracts attention: the second and third most common neuropathology after AD, Parkinson's disease and amyotrophic lateral sclerosis, are also possibly spread throughout the body in a prion-like manner. And there are a number of studies about this [51, 52]. Thus, clusters of alpha-synuclein, the main participant in Parkinson's disease, as described in a recent work in 2019, first appear in the intestine and then move through other tissues like prions. Moreover, these clusters in the gastrointestinal tract can be detected, according to scientists, 20 years before the onset of clinical symptoms of Parkinson's disease. “ In this new study, we found out exactly how the disease can spread from the intestines of people. We probably will not be able to develop effective treatments that will stop the disease without knowing where it starts and how it spreads - so this is an important step in our study of pathology. Parkinson's disease is a complex disease that we are still trying to understand. However, thanks to this study and a similar study in the USA, which recently led to the same result using mice, it is suggested that the disease begins in the intestines of patients, ”says one of the authors of the work, P. Borghammer [53].
Interesting data on similar "related" mechanisms of the pathogenesis of Alzheimer's and Parkinson's diseases were obtained by Z. Zhang et al. They showed that the same enzyme, asparagine endopeptidase (AEP), cleaves both the tau protein in AD and alpha-synuclein in PD, which leads to their aggregation and neurotoxicity [54].
It is quite obvious that the prion-like distribution of the three main neurodegenerative pathologies can hardly be an accident. Although the possibility of infection from the outside is shown today only for Alzheimer's disease. In 2019, American scientists, J. Weickenmeier et al., Were able to bring the theoretical base to the prion hypothesis for these three neuropathologies. They created a physical model for them that explains the prion-like features of neurodegeneration in Alzheimer's, Parkinson’s and amyotrophic lateral sclerosis: “ Our results show that improperly folded proteins in various neurodegenerative diseases grow and spread in accordance with the universal law, which follows the basic physical principles of nonlinear reaction and anisotropic diffusion. Our results confirm the concept of the general fundamental principle of the pathogenesis of a wide range of neurodegenerative disorders - prion paradigm . " Fans of complex formulas and equations will be able to enjoy reading this article [55].
Well, in addition to prions, we will tell you about one more discovered interesting feature of the brain of people with AD. As shown by British scientists, Shelley J. Allen et al., The brain tissue of patients with AD contains an abnormally large number of microbes skewed towards P. acnes . " P. acnes is a commensal gram-positive component of the microflora of the human skin and mouth, preferring anaerobic growth conditions, and it is becoming increasingly apparent that it is a significant opportunistic microorganism. This is most often associated with postoperative lesions and implanted prostheses, as well as chronic diseases such as inflammation of the lumbar, endocarditis, sarcoidosis, and intracranial lesions. Recently, the presence of proteobacteria and actinobacteria (containing Propionibacteriaceae) in mo n, affected both normal and multiple sclerosis; thus, the normal brain has germs, consisting mainly of Proteobacteria and Actinobacteria Our data suggest that Actinobacteria (P. acnes) increases in the brain in AD brain damage proteobacteria ability.. P. acnes nonspecifically stimulate the innate immune system is well-documented "- the authors of [56] write.
“Nutritional" hormones in the pathogenesis of AD
Also in recent work, scientists have found that the hunger hormone ghrelin may be involved in the development of Alzheimer's disease. Ghrelin receptors have recently been discovered in the hippocampus. The hippocampus is an area of the brain that is important for learning, memory and emotions. In AD, this is one of the first areas where cell death and damage due to the formation of amyloid beta accumulations occurs. In a healthy hippocampus, ghrelin binds to its receptor, GHSR1α. This then mediates the activation of the dopamine receptor D1 (DRD1). Modulation of DRD1 by GHSR1α is critical for the function of the hippocampal synapses and synaptic reorganization via the Gαq-Ca 2+ signaling pathway, which is central to memory formation.
The role of GHSR1α in the synaptic function of the hippocampus has allowed scientists to suggest that receptor dysfunction may contribute to the synaptic deficit in the hippocampus observed in AD.
Researchers at the University of Dallas studied this issue using brain samples from people suffering from AD and an animal model from AD. The hypothesis of scientists was that dissociation (i.e. separation) between ghrelin and dopamine receptors may be the very factor that affects cognitive ability in patients with Alzheimer's disease.
Scientists have been able to find that amyloid β inhibits the activation of GHSR1α. Which in turn disrupts the GHSR1α-mediated activation of DRD1 in the hippocampus of people with AD. In an animal model, researchers injected two mice with AD activating the ghrelin and dopamine receptors in the hippocampus, MK0677 and SKF8129. This combination prevented inhibition of the GHSRr1α receptor by β-amyloid, mitigating synaptic damage to the hippocampus and improving learning and memory of rodents [25].
Characteristically, ghrelin is not the first “food" hormone that can be associated with cognitive impairment and Alzheimer's. Prior to this, they discovered the effect of insulin with resistance to it, even calling Alzheimer's type 3 diabetes.
As the available data show, AD can be a slowly progressive metabolic disease of the brain. And numerous studies demonstrate a complex relationship between metabolic syndrome (MetS) and AD. People with diabetes and obesity have a higher risk of developing AD. At the same time, patients with AD often develop hyperglycemia and insulin resistance (IR). IR as a violation of insulin signaling is a common characteristic of both MetS and AD. And, according to scientists, it is a key link between the two diseases. Insulin signaling regulates Aβ and tau levels, and Aβ has a negative effect on insulin signaling. Therefore, insulin signaling dysfunction can enhance the pathology of Aβ and tau, and increased production of Aβ can further exacerbate IR.The accumulated data also indicate that AD is closely related to dysfunction of both insulin signaling and glucose metabolism in the brain, which prompts some researchers to attribute AD to type 3 diabetes or to an insulin-resistant state of the brain [26, 27].
Insulin is secreted by the beta cells of the pancreas and enters the central nervous system, crossing the blood-brain barrier in a controlled and saturated manner. The synthesis of insulin in the brain itself is still a matter of discussion [28]. Insulin receptors (InsRs) are widely expressed in the brain, including the olfactory bulb, cerebral cortex, hippocampus, hypothalamus, and amygdala [29]. InsRs are more concentrated in neurons compared to glial cells [30].
The transmission of insulin signals to the brain plays an important role in regulating food intake, body weight, reproduction, learning and memory. Intranasal administration of insulin improves working memory in both human and animal studies [31]. In addition, the levels of InsR mRNA and protein increase in the CA1 region of the hippocampus during the formation of short-term memory [32]. This suggests that the sensitivity of neurons to insulin may be increased during training.
Impaired insulin signaling makes neurons more vulnerable to metabolic stress, accelerating neuronal dysfunction. Defective insulin signaling is associated with decreased cognitive abilities and the development of dementia, including AD [33]. The deterioration of cognitive abilities in diabetes and AD is associated with a decrease in InsR expression and insulin level in cerebrospinal fluid (CSF) [34, 35]. Decreased insulin signaling, including altered kinase activity and IRS expression, with AD worsens with disease progression [36,37]. And increased phosphorylation of IRS-1, a key IR factor, occurs in the brain with AD [38]. Interestingly, the brain regions with the highest InsR density, such as the hippocampus and temporal lobe, are also the main targets of neurodegeneration in AD [39, 40]. Therefore, impaired insulin signaling,caused by IR can have a big impact on cognitive decline and the development of AD.
Several diabetes treatments that enhance insulin signaling are being tested for therapeutic potential in AD and dementia. Although the results of clinical trials of TZD have been disappointing, intranasal insulin and GLP-1 analogues are still actively used as a potential treatment for AD and have shown some promising results. Intranasal insulin, however, is only effective in the early stages of AD. In addition, exenatide and liraglutide are still in the early stages of therapeutic development, and large clinical trials are currently underway [41].
But that is not all.It turns out that in addition to ghrelin and insulin, two more food hormones, among adipokines (adipose tissue hormones), leptin and adiponectin can participate in Alzheimer's.
There are recent studies from this year:
Obesity as a risk factor for Alzheimer's disease: the effects of leptin and glutamate.
The role of leptin and adiponectin in the cognitive decline associated with obesity and Alzheimer's disease.
Earlier work, 2016 and 2018:
Leptin dysfunction and Alzheimer's disease: data from cell studies, animal and human studies.
Hepocampus leptin regulation and its role in Alzheimer's disease.
There is increasing evidence that leptin has cognitively stimulating properties, as it is involved in the signaling pathways underlying hippocampal learning and memory. However, a significant decrease in the ability of leptin to regulate the synaptic function of the hippocampus occurs with age, and dysfunctions in the leptin system are associated with an increased risk of developing Alzheimer's disease.
www.ncbi.nlm.nih.gov/pubmed/28987937
Another hormone associated with adipose tissue and inflammation, resistin, plays an important role in inflammation of the hypothalamus and hormonal regulation: The
molecular mechanisms underlying obesity-induced inflammation of the hypothalamus and insulin resistance: a key role of the resistin / TLR4 pathway
www.ncbi.nlm.nih.gov/pmc/articles/PMC6418006
In the end, what happens? A number of “food hormones” (ghrelin, insulin, resistin, leptin, adiponectin), the regulation of which is disturbed during aging, can one way or another participate in neurodegenerative processes.
Therefore, it is not at all accidental that antidiabetic drugs, metformin, and others undergo preclinical studies with a view to counteracting age-related neurodegeneration.
Antidiabetic drugs for Alzheimer's disease: mechanisms of action and future prospects
Well, at the very end we’ll briefly say how a violation of sleep regimen and quality can be associated with the development of AD. In 2018, N. Volkow et al. Published in PNAS, in which they showed that even one sleepless night can increase the accumulation of amyloid beta in the brain [57].
And in 2019, scientists from the University of Washington published a study showing that a decrease in deep sleep is associated with early signs of Alzheimer's disease [58].
Summarize.The most common age-related neurodegenerative pathologies show the extremely complex nature of their pathogenesis. To understand which and put into a single picture, we are not yet able to. There are no effective medicines against them yet and it is not even clear on what basis they should be created. Therefore, what a person can do now is to lead a healthy lifestyle and support, to the extent possible, work and research directed against aging in general and age-related neurodegeneration in particular.
The review was prepared by: M. Batin, A. Rzheshevsky.
Bibliography
1.Liu, CC, Liu, CC, Kanekiyo, T., Xu, H., and Bu, G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013; 9: 106–118
2. Penke, B., Bogar, F., and Fulop, L. Beta-amyloid and the Pathomechanisms of Alzheimer's disease: a comprehensive view. Molecules (Basel, Switzerland). 2017; 22: 10
3. Le Page, A., Dupuis, G., Frost, EH, Larbi, A., Pawelec, G., Witkowski, JM et al. Role of the peripheral innate immune system in the development of Alzheimer's disease. Exp Gerontol. 2018; 107: 59–66
4. Bronzuoli, MR, Iacomino, A., Steardo, L., and Scuderi, C. Targeting neuroinflammation in Alzheimer's disease. Journal of inflammation research. 2016; 9: 199–208
5. Kagan, BL, Jang, H., Capone, R., Teran Arce, F., Ramachandran, S., Lal, R. et al. Antimicrobial properties of amyloid peptides. Mol Pharm. 2011; 9: 708–717
6. Jadiya P, Kolmetzky DW, Tomar D, Di Meco A, Lombardi AA, Lambert JP, Luongo TS, Ludtmann MH, Praticò D, Elrod JW. Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer's disease. Nat Commun. 2019 Aug 29;10(1):3885.
7. Temple Scientists Identify Promising New Target to Combat Alzheimer's Disease. www.templehealth.org/about/news/temple-scientists-identify-promising-new-target-to-combat-alzheimers-disease
8. Clemente, JC, Ursell, LK, Parfrey, LW & Knight, R. The impact of the gut microbiota on human health: an integrative view. Cell 148, 1258–1270 (2012).
9. Fung, TC, Olson, CA & Hsiao, EY Interactions between the microbiota, immune and nervous systems in health and disease. Nat. Neurosci. 20, 145–155 (2017).
10.Scheperjans, F. et al. Gut microbiota are related to Parkinson's disease and clinical phenotype. Mov. Disord. 30, 350–358 (2015).
11.Keshavarzian, A. et al. Colonic bacterial composition in Parkinson's disease. Mov. Disord. 30, 1351–1360 (2015).
12. Cattaneo, A. et al. Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol. Aging 49, 60–68 (2017).
13. Minter, MR et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer's disease. Sci. Rep. 6, 1–12 (2016).
14. Harach, T. et al. Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci. Rep. 7, 41802 (2017).
15. Morris, MC and Tangney, CC Dietary fat composition and dementia risk. Neurobiol Aging. 2014; 35: S59–S64
16. Gu, Y. and Scarmeas, N. Dietary patterns in Alzheimer's disease and cognitive aging. Curr Alzheimer Res. 2011; 8: 510–519.
17. Chianese, R., Coccurello, R., Viggiano, A., Scafuro, M., Fiore, M., Coppola, G. et al. Impact of dietary fats on brain functions. Curr Neuropharmacol. 2018; 16: 1059–1085
18. Bach-Faig, A., Berry, EM, Lairon, D., Reguant, J., Trichopoulou, A., Dernini, S. et al. Mediterranean diet pyramid today. Science and cultural updates. Public Health Nutr. 2011; 14: 2274–2284
19. Willett, WC, Sacks, F., Trichopoulou, A., Drescher, G., Ferro-Luzzi, A., Helsing, E. et al. Mediterranean diet pyramid: a cultural model for healthy eating. (Suppl)Am J Clin Nutr. 1995; 61: 1402S–1406S
20. Tosti, V., Bertozzi, B., and Fontana, L. Health benefits of the Mediterranean diet: metabolic and molecular mechanisms. J Gerontol A Biol Sci Med Sci. 2018; 73: 318–326
21. Sofi, F., Abbate, R., Gensini, GF, and Casini, A. Accruing evidence on benefits of adherence to the Mediterranean diet on health: an updated systematic review and meta-analysis. Am J Clin Nutr. 2010; 92: 1189–1196
22. David, LA, Maurice, CF, Carmody, RN, Gootenberg, DB, Button, JE, Wolfe, BE et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014; 505: 559–563
23. Ma, D., Wang, AC, Parikh, I., Green, SJ, Hoffman, JD, Chlipala, G. et al. Ketogenic diet enhances neurovascular function with altered gut microbiome in young healthy mice. Sci Rep. 2018; 8: 6670
24. Nagpal R1, Neth BJ2, Wang S1, Craft S3, Yadav H. Modified Mediterranean-ketogenic diet modulates gut microbiome and short-chain fatty acids in association with Alzheimer's disease markers in subjects with mild cognitive impairment. EBioMedicine. 2019 Aug 30. pii: S2352-3964(19)30554-7.
25. Tian J et al. Disrupted hippocampal growth hormone secretagogue receptor 1α interaction with dopamine receptor D1 plays a role in Alzheimer's disease. Sci Transl Med. 2019 Aug 14;11(505). pii: eaav6278.
26. Frisardi V, Solfrizzi V, Capurso C, Imbimbo BP, Vendemiale G, Seripa D et al. Is insulin resistant brain state a central feature of the metabolic-cognitive syndrome? J Alzheimers Dis 2010; 21: 57–63.
27. Lester-Coll N, Rivera EJ, Soscia SJ, Doiron K, Wands JR, de la Monte SM. Intracerebral streptozotocin model of type 3 diabetes: relevance to sporadic Alzheimer's disease. J Alzheimers Dis 2006; 9: 13–33.
28. Baskin DG, Figlewicz DP, Woods SC, Porte D Jr ., Dorsa DM. Insulin in the brain. Annu Rev Physiol 1987; 49: 335–347.
29. Unger JW, Livingston JN, Moss AM. Insulin receptors in the central nervous system: localization, signalling mechanisms and functional aspects. Prog Neurobiol 1991; 36: 343–362.
30.van der Heide LP, Ramakers GM, Smidt MP. Insulin signaling in the central nervous system: learning to survive. Prog Neurobiol 2006; 79: 205–221.
31. Benedict C, Frey WH 2nd, Schioth HB, Schultes B, Born J, Hallschmid M. Intranasal insulin as a therapeutic option in the treatment of cognitive impairments. Exp Gerontol 2011; 46: 112–115.
32. Zhao W, Chen H, Xu H, Moore E, Meiri N, Quon MJ et al. Brain insulin receptors and spatial memory. Correlated changes in gene expression, tyrosine phosphorylation, and signaling molecules in the hippocampus of water maze trained rats. J Biol Chem 1999; 274: 34893–34902.
33.de la Monte SM. Insulin resistance and Alzheimer's disease. BMB Rep 2009; 42: 475–481.
34. Duarte AI, Moreira PI, Oliveira CR. Insulin in central nervous system: more than just a peripheral hormone. Journal of aging research 2012; 2012: 384017.
35. Moloney AM, Griffin RJ, Timmons S, O'Connor R, Ravid R, O'Neill C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer's disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol Aging 2010; 31: 224–243.
36. Bosco D, Fava A, Plastino M, Montalcini T, Pujia A. Possible implications of insulin resistance and glucose metabolism in Alzheimer's disease pathogenesis. J Cell Mol Med 2011; 15: 1807–1821.
36. Li L, Holscher C. Common pathological processes in Alzheimer disease and type 2 diabetes: a review. Brain Res Rev 2007; 56: 384–402.
37. Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A et al. Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest 2012; 122: 1316–1338.
38. de la Monte SM, Wands JR. Review of insulin and insulin-like growth factor expression, signaling, and malfunction in the central nervous system: relevance to Alzheimer's disease. J Alzheimers Dis 2005; 7: 45–61.
39. Freude S, Schilbach K, Schubert M. The role of IGF-1 receptor and insulin receptor signaling for the pathogenesis of Alzheimer's disease: from model organisms to human disease. Curr Alzheimer Res 2009; 6: 213–223.
40. Gammeltoft S, Fehlmann M, Van Obberghen E. Insulin receptors in the mammalian central nervous system: binding characteristics and subunit structure. Biochimie 1985; 67: 1147–1153.
41. Bhumsoo Kim, Eva L Feldman. Insulin resistance as a key link for the increased risk of cognitive impairment in the metabolic syndrome Exp Mol Med. 2015 Mar; 47(3): e149. Published online 2015 Mar 13.
42. Jing Sun et al. Fecal microbiota transplantation alleviated Alzheimer's disease-like pathogenesis in APP/PS1 transgenic mice. Transl Psychiatry. 2019; 9: 189.
43. Shalini Elangovan, Thomas J Borody, Damian Holsinger. Fecal Microbiota Transplantation Decreases Amyloid Load and Improves Cognition in Alzheimer's. BioRxiv Jul 1, 2019.
44. Kim M, Kim Y, Choi H, et al Transfer of a healthy microbiota reduces amyloid and tau pathology in an Alzheimer's disease animal model. Gut. Published Online First: 30 August 2019. doi: 10.1136/gutjnl-2018-317431.
45. Shih-Yi Lin et al. Association of Transfusion With Risks of Dementia or Alzheimer's Disease: A Population-Based Cohort Study Front Psychiatry. 2019; 10: 571.
46. Jaunmuktane Z et al. Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nature. 2015 Sep 10;525(7568):247-50.
47. Purro SA et al. Transmission of amyloid-β protein pathology from cadaveric pituitary growth hormone. Nature. 2018 Dec;564(7736):415-419. doi: 10.1038/s41586-018-0790-y. Epub 2018 Dec 13.
48. Jaunmuktane Z et al. Evidence of amyloid-β cerebral amyloid angiopathy transmission through neurosurgery. Acta Neuropathol. 2018 May;135(5):671-679.
49. Bu XL et al. Blood-derived amyloid-β protein induces Alzheimer's disease pathologies. Mol Psychiatry. 2018 Sep;23(9):1948-1956.
50. Atsushi Aoyagi, Carlo Condello, Jan Stöhr, Weizhou Yue, Brianna M. Rivera, Joanne C. Lee, Amanda L. Woerman, Glenda Halliday, Sjoerd Van Duinen, Martin Ingelsson, Lars Lannfelt, Caroline Graff, Thomas D. Bird, C. Dirk Keene, William W. Seeley, William F. Degrado, and Stanley B. Prusiner. Aβ and tau prion-like activities decline with longevity in the Alzheimer's disease human brain. Science Translational Medicine, 2019.
51. Ayers JI, Cashman NR. Prion-like mechanisms in amyotrophic lateral sclerosis.
Handb Clin Neurol. 2018;153:337-354.
52. Kujawska M, Jodynis-Liebert J.. What is the Evidence That Parkinson's Disease is a Prion Disorder, Which Originates in the Gut? Int J Mol Sci. 2018 Nov 12;19(11). pii: E3573
53.Van Den Berge N et al. Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol. 2019 Jun 26.
54. Zhentao Zhang, Seong Su Kang, Xia Liu, Eun Hee Ahn, Zhaohui Zhang, Li He, P Michael Iuvone, Duc M Duong, Nicholas T Seyfried, Matthew J Benskey, Fredric P Manfredsson, Lingjing Jin, Yi E Sun, Jian-Zhi Wang, Keqiang Ye. Asparagine endopeptidase cleaves α-synuclein and mediates pathologic activities in Parkinson's disease. Nature Structural & Molecular Biology, 2017; DOI: 10.1038/nsmb.3433
55. Johannes Weickenmeier, Mathias Juckerb, Alain Gorielyc, Ellen Kuh. A physics-based model explains the prion-like features of neurodegeneration in Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis Author links open overlay panel. Journal of the Mechanics and Physics of Solids Volume 124, March 2019, Pages 264-281.
56. David C. Emery, Deborah K. Shoemark, Tom E. Batstone, Christy M. Waterfall, Jane A. Coghill, Tanya L. Cerajewska, Maria Davies, Nicola X. West, Shelley J. Allen. 16S rRNA Next Generation Sequencing Analysis Shows Bacteria in Alzheimer's Post-Mortem Brain. Frontiers in Aging Neuroscience, 2017; 9 DOI: 10.3389/fnagi.2017.00195
57. Shokri-Kojori E. et al. β-Amyloid accumulation in the human brain after one night of sleep deprivation. Proc Natl Acad Sci US A. 2018 Apr 24;115(17):4483-4488.
58. Lucey BP, McCullough A, Landsness EC, Toedebusch CD, McLeland JS, Zaza AM, Fagan AM, McCue L, Xiong C, Morris JC, Benzinger TLS, Holtzman DM. Reduced non-rapid eye movement sleep is associated with tau pathology in early Alzheimer's disease. Science Translational Medicine, Jan. 9, 2019
2. Penke, B., Bogar, F., and Fulop, L. Beta-amyloid and the Pathomechanisms of Alzheimer's disease: a comprehensive view. Molecules (Basel, Switzerland). 2017; 22: 10
3. Le Page, A., Dupuis, G., Frost, EH, Larbi, A., Pawelec, G., Witkowski, JM et al. Role of the peripheral innate immune system in the development of Alzheimer's disease. Exp Gerontol. 2018; 107: 59–66
4. Bronzuoli, MR, Iacomino, A., Steardo, L., and Scuderi, C. Targeting neuroinflammation in Alzheimer's disease. Journal of inflammation research. 2016; 9: 199–208
5. Kagan, BL, Jang, H., Capone, R., Teran Arce, F., Ramachandran, S., Lal, R. et al. Antimicrobial properties of amyloid peptides. Mol Pharm. 2011; 9: 708–717
6. Jadiya P, Kolmetzky DW, Tomar D, Di Meco A, Lombardi AA, Lambert JP, Luongo TS, Ludtmann MH, Praticò D, Elrod JW. Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer's disease. Nat Commun. 2019 Aug 29;10(1):3885.
7. Temple Scientists Identify Promising New Target to Combat Alzheimer's Disease. www.templehealth.org/about/news/temple-scientists-identify-promising-new-target-to-combat-alzheimers-disease
8. Clemente, JC, Ursell, LK, Parfrey, LW & Knight, R. The impact of the gut microbiota on human health: an integrative view. Cell 148, 1258–1270 (2012).
9. Fung, TC, Olson, CA & Hsiao, EY Interactions between the microbiota, immune and nervous systems in health and disease. Nat. Neurosci. 20, 145–155 (2017).
10.Scheperjans, F. et al. Gut microbiota are related to Parkinson's disease and clinical phenotype. Mov. Disord. 30, 350–358 (2015).
11.Keshavarzian, A. et al. Colonic bacterial composition in Parkinson's disease. Mov. Disord. 30, 1351–1360 (2015).
12. Cattaneo, A. et al. Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol. Aging 49, 60–68 (2017).
13. Minter, MR et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer's disease. Sci. Rep. 6, 1–12 (2016).
14. Harach, T. et al. Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci. Rep. 7, 41802 (2017).
15. Morris, MC and Tangney, CC Dietary fat composition and dementia risk. Neurobiol Aging. 2014; 35: S59–S64
16. Gu, Y. and Scarmeas, N. Dietary patterns in Alzheimer's disease and cognitive aging. Curr Alzheimer Res. 2011; 8: 510–519.
17. Chianese, R., Coccurello, R., Viggiano, A., Scafuro, M., Fiore, M., Coppola, G. et al. Impact of dietary fats on brain functions. Curr Neuropharmacol. 2018; 16: 1059–1085
18. Bach-Faig, A., Berry, EM, Lairon, D., Reguant, J., Trichopoulou, A., Dernini, S. et al. Mediterranean diet pyramid today. Science and cultural updates. Public Health Nutr. 2011; 14: 2274–2284
19. Willett, WC, Sacks, F., Trichopoulou, A., Drescher, G., Ferro-Luzzi, A., Helsing, E. et al. Mediterranean diet pyramid: a cultural model for healthy eating. (Suppl)Am J Clin Nutr. 1995; 61: 1402S–1406S
20. Tosti, V., Bertozzi, B., and Fontana, L. Health benefits of the Mediterranean diet: metabolic and molecular mechanisms. J Gerontol A Biol Sci Med Sci. 2018; 73: 318–326
21. Sofi, F., Abbate, R., Gensini, GF, and Casini, A. Accruing evidence on benefits of adherence to the Mediterranean diet on health: an updated systematic review and meta-analysis. Am J Clin Nutr. 2010; 92: 1189–1196
22. David, LA, Maurice, CF, Carmody, RN, Gootenberg, DB, Button, JE, Wolfe, BE et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014; 505: 559–563
23. Ma, D., Wang, AC, Parikh, I., Green, SJ, Hoffman, JD, Chlipala, G. et al. Ketogenic diet enhances neurovascular function with altered gut microbiome in young healthy mice. Sci Rep. 2018; 8: 6670
24. Nagpal R1, Neth BJ2, Wang S1, Craft S3, Yadav H. Modified Mediterranean-ketogenic diet modulates gut microbiome and short-chain fatty acids in association with Alzheimer's disease markers in subjects with mild cognitive impairment. EBioMedicine. 2019 Aug 30. pii: S2352-3964(19)30554-7.
25. Tian J et al. Disrupted hippocampal growth hormone secretagogue receptor 1α interaction with dopamine receptor D1 plays a role in Alzheimer's disease. Sci Transl Med. 2019 Aug 14;11(505). pii: eaav6278.
26. Frisardi V, Solfrizzi V, Capurso C, Imbimbo BP, Vendemiale G, Seripa D et al. Is insulin resistant brain state a central feature of the metabolic-cognitive syndrome? J Alzheimers Dis 2010; 21: 57–63.
27. Lester-Coll N, Rivera EJ, Soscia SJ, Doiron K, Wands JR, de la Monte SM. Intracerebral streptozotocin model of type 3 diabetes: relevance to sporadic Alzheimer's disease. J Alzheimers Dis 2006; 9: 13–33.
28. Baskin DG, Figlewicz DP, Woods SC, Porte D Jr ., Dorsa DM. Insulin in the brain. Annu Rev Physiol 1987; 49: 335–347.
29. Unger JW, Livingston JN, Moss AM. Insulin receptors in the central nervous system: localization, signalling mechanisms and functional aspects. Prog Neurobiol 1991; 36: 343–362.
30.van der Heide LP, Ramakers GM, Smidt MP. Insulin signaling in the central nervous system: learning to survive. Prog Neurobiol 2006; 79: 205–221.
31. Benedict C, Frey WH 2nd, Schioth HB, Schultes B, Born J, Hallschmid M. Intranasal insulin as a therapeutic option in the treatment of cognitive impairments. Exp Gerontol 2011; 46: 112–115.
32. Zhao W, Chen H, Xu H, Moore E, Meiri N, Quon MJ et al. Brain insulin receptors and spatial memory. Correlated changes in gene expression, tyrosine phosphorylation, and signaling molecules in the hippocampus of water maze trained rats. J Biol Chem 1999; 274: 34893–34902.
33.de la Monte SM. Insulin resistance and Alzheimer's disease. BMB Rep 2009; 42: 475–481.
34. Duarte AI, Moreira PI, Oliveira CR. Insulin in central nervous system: more than just a peripheral hormone. Journal of aging research 2012; 2012: 384017.
35. Moloney AM, Griffin RJ, Timmons S, O'Connor R, Ravid R, O'Neill C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer's disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol Aging 2010; 31: 224–243.
36. Bosco D, Fava A, Plastino M, Montalcini T, Pujia A. Possible implications of insulin resistance and glucose metabolism in Alzheimer's disease pathogenesis. J Cell Mol Med 2011; 15: 1807–1821.
36. Li L, Holscher C. Common pathological processes in Alzheimer disease and type 2 diabetes: a review. Brain Res Rev 2007; 56: 384–402.
37. Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A et al. Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest 2012; 122: 1316–1338.
38. de la Monte SM, Wands JR. Review of insulin and insulin-like growth factor expression, signaling, and malfunction in the central nervous system: relevance to Alzheimer's disease. J Alzheimers Dis 2005; 7: 45–61.
39. Freude S, Schilbach K, Schubert M. The role of IGF-1 receptor and insulin receptor signaling for the pathogenesis of Alzheimer's disease: from model organisms to human disease. Curr Alzheimer Res 2009; 6: 213–223.
40. Gammeltoft S, Fehlmann M, Van Obberghen E. Insulin receptors in the mammalian central nervous system: binding characteristics and subunit structure. Biochimie 1985; 67: 1147–1153.
41. Bhumsoo Kim, Eva L Feldman. Insulin resistance as a key link for the increased risk of cognitive impairment in the metabolic syndrome Exp Mol Med. 2015 Mar; 47(3): e149. Published online 2015 Mar 13.
42. Jing Sun et al. Fecal microbiota transplantation alleviated Alzheimer's disease-like pathogenesis in APP/PS1 transgenic mice. Transl Psychiatry. 2019; 9: 189.
43. Shalini Elangovan, Thomas J Borody, Damian Holsinger. Fecal Microbiota Transplantation Decreases Amyloid Load and Improves Cognition in Alzheimer's. BioRxiv Jul 1, 2019.
44. Kim M, Kim Y, Choi H, et al Transfer of a healthy microbiota reduces amyloid and tau pathology in an Alzheimer's disease animal model. Gut. Published Online First: 30 August 2019. doi: 10.1136/gutjnl-2018-317431.
45. Shih-Yi Lin et al. Association of Transfusion With Risks of Dementia or Alzheimer's Disease: A Population-Based Cohort Study Front Psychiatry. 2019; 10: 571.
46. Jaunmuktane Z et al. Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nature. 2015 Sep 10;525(7568):247-50.
47. Purro SA et al. Transmission of amyloid-β protein pathology from cadaveric pituitary growth hormone. Nature. 2018 Dec;564(7736):415-419. doi: 10.1038/s41586-018-0790-y. Epub 2018 Dec 13.
48. Jaunmuktane Z et al. Evidence of amyloid-β cerebral amyloid angiopathy transmission through neurosurgery. Acta Neuropathol. 2018 May;135(5):671-679.
49. Bu XL et al. Blood-derived amyloid-β protein induces Alzheimer's disease pathologies. Mol Psychiatry. 2018 Sep;23(9):1948-1956.
50. Atsushi Aoyagi, Carlo Condello, Jan Stöhr, Weizhou Yue, Brianna M. Rivera, Joanne C. Lee, Amanda L. Woerman, Glenda Halliday, Sjoerd Van Duinen, Martin Ingelsson, Lars Lannfelt, Caroline Graff, Thomas D. Bird, C. Dirk Keene, William W. Seeley, William F. Degrado, and Stanley B. Prusiner. Aβ and tau prion-like activities decline with longevity in the Alzheimer's disease human brain. Science Translational Medicine, 2019.
51. Ayers JI, Cashman NR. Prion-like mechanisms in amyotrophic lateral sclerosis.
Handb Clin Neurol. 2018;153:337-354.
52. Kujawska M, Jodynis-Liebert J.. What is the Evidence That Parkinson's Disease is a Prion Disorder, Which Originates in the Gut? Int J Mol Sci. 2018 Nov 12;19(11). pii: E3573
53.Van Den Berge N et al. Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol. 2019 Jun 26.
54. Zhentao Zhang, Seong Su Kang, Xia Liu, Eun Hee Ahn, Zhaohui Zhang, Li He, P Michael Iuvone, Duc M Duong, Nicholas T Seyfried, Matthew J Benskey, Fredric P Manfredsson, Lingjing Jin, Yi E Sun, Jian-Zhi Wang, Keqiang Ye. Asparagine endopeptidase cleaves α-synuclein and mediates pathologic activities in Parkinson's disease. Nature Structural & Molecular Biology, 2017; DOI: 10.1038/nsmb.3433
55. Johannes Weickenmeier, Mathias Juckerb, Alain Gorielyc, Ellen Kuh. A physics-based model explains the prion-like features of neurodegeneration in Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis Author links open overlay panel. Journal of the Mechanics and Physics of Solids Volume 124, March 2019, Pages 264-281.
56. David C. Emery, Deborah K. Shoemark, Tom E. Batstone, Christy M. Waterfall, Jane A. Coghill, Tanya L. Cerajewska, Maria Davies, Nicola X. West, Shelley J. Allen. 16S rRNA Next Generation Sequencing Analysis Shows Bacteria in Alzheimer's Post-Mortem Brain. Frontiers in Aging Neuroscience, 2017; 9 DOI: 10.3389/fnagi.2017.00195
57. Shokri-Kojori E. et al. β-Amyloid accumulation in the human brain after one night of sleep deprivation. Proc Natl Acad Sci US A. 2018 Apr 24;115(17):4483-4488.
58. Lucey BP, McCullough A, Landsness EC, Toedebusch CD, McLeland JS, Zaza AM, Fagan AM, McCue L, Xiong C, Morris JC, Benzinger TLS, Holtzman DM. Reduced non-rapid eye movement sleep is associated with tau pathology in early Alzheimer's disease. Science Translational Medicine, Jan. 9, 2019
All Articles