Paul-Jean Letourneauの投稿「 MathematicaおよびHadoopLinkを使用したゲノムの検索 」の翻訳。
記事のコードはここからダウンロードできます 。
注:この投稿はMathematicaでのBig Data Arrays with HadoopLinkの投稿の続きとして書かれています。
翻訳者注 :この記事の著者は、 遺伝子という用語の下で、生物の構造要素の遺伝子の全体を理解しています。 これは、特定の種の遺伝子のセット全体(Ridley、M.(2006)。Genome。New York、NY:Harper Perennial)、またはセル( http://www.genome.gov/Glossary/index.cfm?id=90 )。 この投稿では、著者のプレゼンテーションを使用します。
以前の投稿で 、 HadoopLinkパッケージを使用してMathematicaで MapReduceアルゴリズム( wiki )を記述する方法を説明しました。 次に、もう少し深く掘り下げて、より深刻なMapReduceアルゴリズムを作成しましょう。
Wolfram | Alphaでの楽しいゲノミクスの機会について以前に書いた。 興味がある場合は、特定のDNA配列を人間のゲノムで検索することもできます。 生物学者は、多くの場合、どの動物がこのフラグメントに属しているか、またはどの染色体から属しているかを判断するために、研究室で見つけたDNAフラグメントの場所を見つける必要があります。 HadoopLinkを使用して、ゲノム検索エンジンを作成しましょう!
前と同様に、 HadoopLinkパッケージをダウンロードします。
メインのHadoopノードでリンクを設定します:

このアイデアを説明するために、 GenomeDataから小さなヒトミトコンドリアゲノムを取得します。

まず、ゲノムを別々のベース(A、T、C、G)に分割します。

キーと値のペア(k1、v1)になります。 値は、ゲノムの各塩基の初期位置です。
{k1、v1} = {base、position}
つまり、ゲノムのインデックスを作成しただけです。
このインデックスをHadoop分散ファイルシステム (HDFS)にエクスポートします。
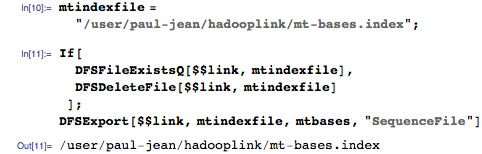
クエリには、11塩基のシーケンスを使用しますが、そのうち11塩基がゲノムに含まれていることがわかります。

アルゴリズムのタスクを複雑にするために、多数の繰り返しを含むシーケンスを使用します。
ゲノム検索エンジンは、位置515をこのリクエストに返す必要があります。
今、 mapperとreducerが必要です。
前半で述べたように、 マッパーはキーと値のペアを取得し(ステップ1)、別のペアを生成します(ステップ2)。

Mapperは、入力値としてゲノムインデックスからベースを取得し、ベースインデックスが見つかったクエリ内の各場所のキーと値のペアを生成します(理由はすぐにわかります)。
(1)入力: {k1、v1} = {ベースインデックス、ゲノム内の位置}

出力のキーはゲノム内の位置であり、出力の値はリクエスト内の位置です。
(2)結論: {k2、v2} = {ゲノム内の位置、リクエスト内の位置}
ゲノム内の位置とリクエスト内の位置の違いは何ですか? クエリ内の位置は、クエリ内の基本位置です。一方、ゲノム内の位置は、ゲノム全体内の位置です。
たとえば、 マッパーが位置517にベースAを持つキーと値のペアを取得するとします。
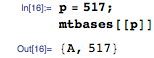
要求されたGCACACACACAAシーケンスのベースAに対する要求の位置は、3、5、7、9、および11です。
強調表示された位置のシーケンスは次のとおりです。

マッパーには、要求されたシーケンスとともに1つのインデックスベースを持つ1つのキーと値のペアのみがあります。 彼は、比較のために残りのゲノムを持っていないため、位置517のベースAでクエリを作成するためのすべての可能な方法を見つける必要があります。

ここで、色はクエリ内の各A(水平)に対応し、ゲノム内の結果の位置(垂直)に対応しています。 たとえば、クエリの3番目のベース(緑色で表示)のAを使用します。 Aから位置517に挿入された場合、要求された配列はゲノムの位置515から始まります(517-3 + 1 = 515)(緑色でも表示)。
同様に、赤(クエリの5番目の位置)で強調表示された塩基は、要求された配列をゲノムの位置513から開始します(これも赤で表示されます)。 ゲノム内に511の位置を持つクエリの7番目の位置(紫色)、ゲノム内に509の位置を持つクエリの9番目の位置(オレンジ)、ゲノム内に507の位置を持つクエリの11番目の位置(茶色)についても同じです。
これらの配置の1つだけが正しいです。 この場合、要求の3番目の位置(緑色)に対して、要求された配列をゲノムに統合できます。 ただし、 マッパーはこれを認識していないため、可能な一致をすべて考慮します。
そのため、 リデューサーはキーを収集し、ゲノム内の同じ位置に対応するすべてのベースを収集します。
入力 : {k2、{v2 ...}} = {ゲノム内の位置、{クエリ内の位置...}}
ゲノム内のこの位置について、値がクエリの位置から完全なシーケンスを形成する場合、 reducerはこれらのケースを探します。

レデューサーが完全に一致する場合、ゲノムの位置を示します:
結論 : {k3、v3} = {リクエストされた配列、ゲノム内の位置}
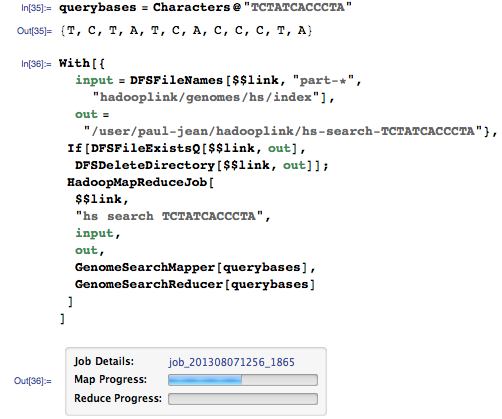
次に、GCACACACACAシーケンスについてゲノム検索エンジンに問い合わせてみましょう。
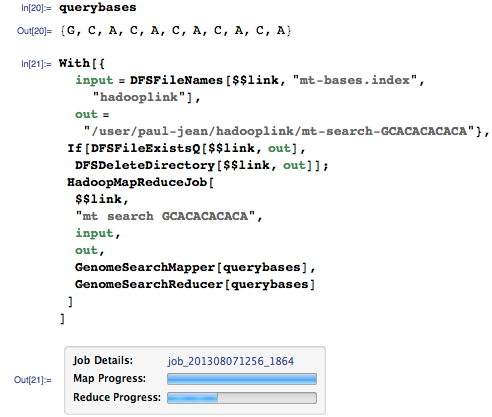
( HadoopMapReduceJob関数の説明については、 パート1を参照してください。 )
そして、HDFSからゲノムマッピングをインポートします。
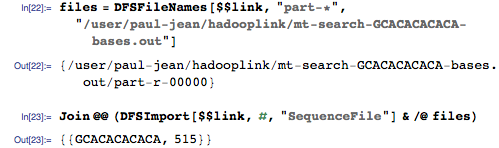
ゲノム内の対応する位置は515であり、これが正解です! ゲノム検索エンジンが機能します!
次に、ゲノム内の2つの異なる位置に対応する必要があるクエリで検索を実行しましょう。

このクエリは、アイテム10と2277に一致する必要があります。
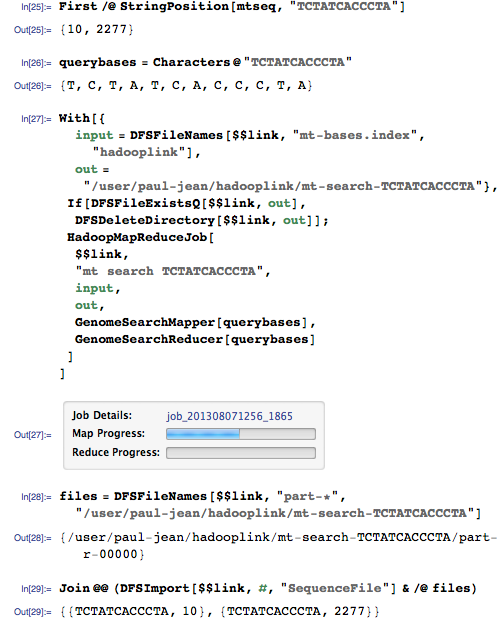
はい、彼は両方の試合を見つけました!
それでは、ヒトゲノム全体についてスケールアウトしましょう。 最初のステップは、今回はミトコンドリアのインデックスだけでなく、ゲノム全体のインデックスを作成することです。 これを行うために、 状態サーバーからテキストファイルとして人間のゲノム全体をダウンロードし、HDFSにインポートしました。

生の染色体配列を含む染色体ごとに1つのテキストファイルが見つかりました。
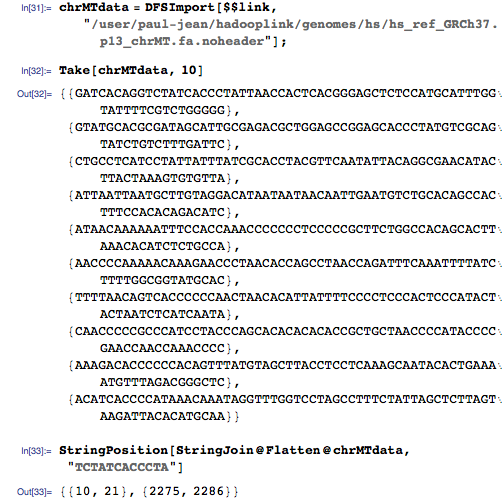
次に、 MapReduceを適用して、HDFSのインデックスにキーと値のペアを作成します。これは次のようになります。
[hs_ref_GRCh37.p13_alts.fa、121] G
[hs_ref_GRCh37.p13_alts.fa、122] A
[hs_ref_GRCh37.p13_alts.fa、123] A
[hs_ref_GRCh37.p13_alts.fa、124] T
[hs_ref_GRCh37.p13_alts.fa、125] T
[hs_ref_GRCh37.p13_alts.fa、126] C
[hs_ref_GRCh37.p13_alts.fa、127] A
[hs_ref_GRCh37.p13_alts.fa、128] G
[hs_ref_GRCh37.p13_alts.fa、129] C
[hs_ref_GRCh37.p13_alts.fa、130] T
前の例とのわずかな違いは、現在のキーは{染色体、ゲノム内の位置}であり、値はこの位置に基づいていることです。 だから今、染色体が鍵になります。 マッパーを少し変更して、新しいキー表現で動作するようにしました。

レデューサーは変更されませんでした。
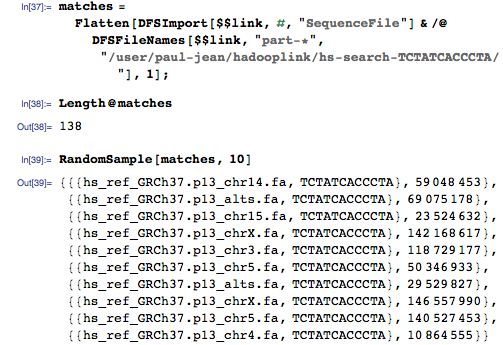
同じシーケンスの検索を実行しましょう:
今回は、ゲノム全体の一致を取得します。
また、アルゴリズムを改善することもできます。 正確な一致ではなく不正確な一致を見つけるのはどうですか? レデューサーをわずかに変更して、一致するリクエストの量を示します。

これはゲノムを検索する最も効率的な方法ではありませんが、 MapReduceに基づいたアルゴリズムのプロトタイプを作成してMathematicaで実行することがいかに簡単かを示しています。 詳細に興味がある場合は、 最近の投稿をご覧になることをお勧めします。 そして、 HadoopLink GitHub Repoに興味のある質問にお答えします !