
ウイルス粒子インフルエンザA / H1N1の成分
ウイルス粒子は、2つの基本的な問題を解決する分子メカニズムです。 第一に、粒子はウイルスゲノムのパッケージングと破壊的な環境要因からの保護を確保する必要がありますが、ウイルスはそれが集まった細胞から感染可能な細胞に移動します。 第二に、粒子は感染細胞に付着し、ウイルスゲノムと関連分子を内部に送達して、新しい生殖サイクルを開始できる必要があります。 タスクはそれほど多くないため、ウイルスは、まれな例外を除いて、構造的に非常に経済的です。
特に、ほとんどのウイルスのゲノムは小さく、あまり多くのタンパク質をエンコードしません。多くの場合、この数は10未満です。さらに、ウイルスは、細胞に同じタイプの多数のタンパク質を合成させ、そこからウイルスエンベロープ、キャプシドが組み立てられます。 したがって、ウイルス粒子は通常、コンストラクターの一部として互いに結合する多数の同一の要素で構成され、しばしば規則的で対称的な構造を形成します。 そのため、すべてのバイラルパッケージまたはそのフラグメントがスパイラルまたは正二十面体の形をしているわけではありませんが、非常に多くのパッケージがあります。

二十面体対称性を持つウイルスカプシドの例。 右下隅のバクトリロドプシン分子-比較用。 ( レビューの図 )。
ウイルスモデルを構築するには、一般構造の個々のタンパク質がどのように配置され、どのように互いに結合してこの構造を形成するかを知ることが重要です。 現代科学には、これらの質問に答えることができるさまざまな方法がありますが、残念ながら、どのアプローチも普遍的ではなく、科学的に信頼できるウイルスのモデルを原子粒度で作成するときに直面するタスクの一部のみを解決します。
タンパク質:構造に関する情報をどのように受け取り、保存し、表示しますか?
タンパク質は、連続して一緒にリンクされたモノマー-アミノ酸からなるポリマー分子であることを思い出してください。 水溶液では、タンパク質は通常、複雑な3次元の小球(ほぼルービックスネークパズルのように)に折り畳まれ、その形状はアミノ酸組成や他のいくつかの要因に依存します。 これらの小球の空間構造は、主にX線回折分析とNMR分光法によって決定されます。 また、電子顕微鏡は最近この問題に取り組むことを可能にしました。
一般に、分子の空間構造を決定する方法は複雑であり、一連の制限があります;したがって、すべてのウイルス蛋白質から完全に記述されています。 そのため、X線分析では、X線が透過する結晶の存在を想定しています。 結晶の原子はX線回折を引き起こし、その画像から結晶中の電子密度の分布を推定することができ、このデータから特定の原子の配置をすでに復元することができます。 この方法では、最大1オングストローム(0.1 nm)を超える分解能が得られますが、タンパク質の場合、すべての結晶を結晶化できるわけではないという問題があります。 タンパク質が膜内に柔軟な可動性または固定フラグメントを持っている場合、これは特に難しいことがわかります。
NMR分光法は核磁気共鳴の現象に基づいており、溶液中のタンパク質の構造を記述することができます。 このアプローチは、分子内の原子の可能な位置のセットを明らかにし、以前の方法とは対照的に、そのセクションのいずれかの柔軟性の程度を評価することを可能にします。 しかし、NMR分光法は比較的小さな分子に対してのみ有効です。なぜなら、大きなタンパク質は非常に多くのノイズを生成するからです。
電子顕微鏡検査により、大きな分子複合体の構造を記述することができます。これは、ウイルスに関して非常に有用です。 多くの対称構造では、さまざまな角度で大量の画像を取得し、それを分析して3次元画像を再作成できます。 個々のオブジェクトについて、異なるタイプの電子顕微鏡(最大4-5オングストローム)を使用した結果として得られる解像度は、X線回折分析の解像度よりもそれほど悪くはありませんが、通常、完全な情報を得るために異なるアプローチを組み合わせて、たとえば個々のタンパク質の構造を「適合」する必要があります電子顕微鏡で得られた電子密度マップ。

HIVエンベロープタンパク質の三量体(分子の赤と青の断片)の構造は、9Åの解像度の低温電子顕微鏡で得られた電子密度マップに刻まれたこのタンパク質に対する抗体の一部(緑と黄色の断片)と複合体を形成しています。 記事「 三量体HIV-1エンベロープ糖タンパク質活性化の構造メカニズム」から。
以前の投稿で書いたように、結果の構造は体系化され、 Protein Data Bankデータベースに保存されます。 同時に、原子座標は* .pdb形式で記録され、これらのデータを視覚化してそのような構造を操作できるようにするプログラムのセットがすべてあります。 それらの中で、例えば、 VMD 、 キメラ 、 PyMol 、および他の何十もの 。
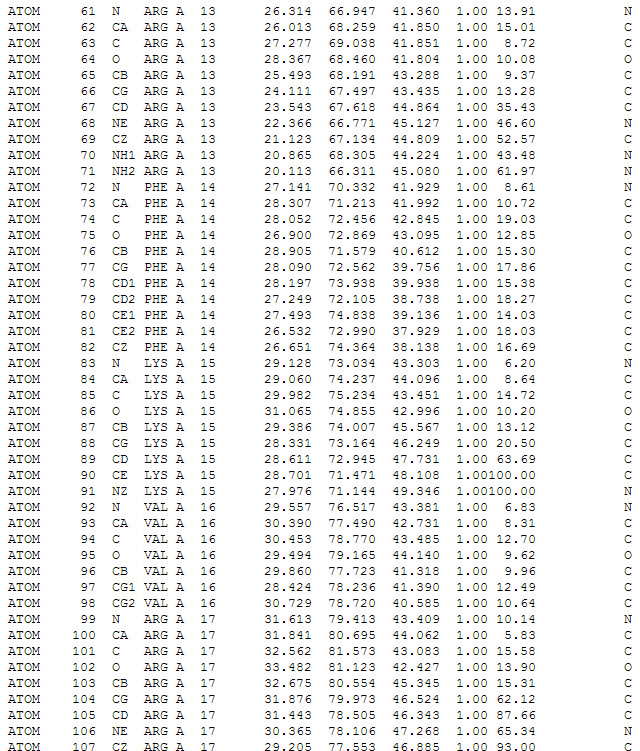
* .pdb形式のファイルのテキスト表示のスクリーンショット。 タンパク質のアミノ酸の個々の原子の座標が記述されています。
プログラムは、いくつかの方法でタンパク質を表示できます。 原子のファンデルワールス半径に対応するさまざまな直径の球による原子の単純な表示に加えて、個々の結合、分子の表面、およびリボンに似た構造を使用してアミノ酸鎖の曲がりを表示することができます( リボン図 ) アルファヘリックスを形成します。ここで、 ベータ層と非構造化領域が形成されます。

キメラプログラムでインフルエンザウイルスの血球凝集素の外側部分の構造を視覚化するためのさまざまなオプション。
軽Asとして、科学者が通常作業しているプログラムで、個々の分子またはタンパク質複合体を視覚化すると、ほとんどの場合美的観点から非常に原始的な結果しか得られません(たとえば、 VMDプログラムのスクリーンショットを見るだけで十分です)。 プロのデザイナーやコンピューターの3次元グラフィックスの専門家が使用するプログラムに分子のモデルをインポートすると、根本的に幅広い可能性が開かれます。 これらのプログラムと、レンダリング品質を向上させるプラグインを組み合わせることで、本当に興味深く魅力的な視覚化を実現できます。 これについては、今後の投稿で詳しく説明します。 とりあえず、例を挙げてください。

免疫グロブリンG分子画像 。
分子モデリング
不足しているタンパク質構造は、ウイルス粒子の完全なモデルを作成するために私たちがしなければならないことを予測しようとします。 これを行うために、すでに説明した構造に関するデータと、分子の個々の原子間の相互作用を特定の信頼性で計算することを可能にするアルゴリズムに基づいて、多くのコンピューター手法が使用されます。 既知の構造に基づいたモデリングが使用されます。これは、現代の計算能力では、量子力学の原理に基づいて、アミノ酸のみに基づいてタンパク質の空間モデルをまだ構築できないためです。 さらに、非常に多くのタンパク質の折り畳みがすでに決定されており、ほぼすべての新しいタンパク質構造について、PDBバンクにすでに類似体が存在すると考えられていますが、主なことはそれを見つけることです。
30%を超えるアミノ酸残基が同一のタンパク質は、非常に類似した構造を持つことが知られています。 類似のアミノ酸配列と既知の構造を持つタンパク質を見つけ、モデルを構築するためのテンプレートとして使用できます-これはホモロジーモデリングと呼ばれます。 同様のシーケンスを見つけるには、通常BLASTプログラムが使用されます。
ただし、構造が似ている一部のタンパク質は、ランダムに選択された一対のタンパク質とほぼ同じ配列類似性を持っています。 このような場合に適切なテンプレートを見つけるには、Fold認識方法を使用します。 彼らは、さまざまな既知の構造でシミュレートされたタンパク質の配列を「引き出し」、このテンプレートがそれらにどのように適合するかを評価します。 異なるプログラムは異なる評価関数を使用するため、異なる結果が生成されます。 現在、フォールド認識のための単一の最適なアルゴリズムは存在しません。通常、一度に複数のプログラムを使用し、すべての結果に基づいてテンプレートを選択します。 たとえば、テンプレートと同様の機能を持つタンパク質を使用できます。
複数のテンプレートを一度に使用してモデルを組み立て、それらを最適な方法で組み合わせることができる方法があります。 それらのベストはI-Tasserと呼ばれます。 プログラムの作成者はそれを最高のものとして発表しませんでした-数年前から、I-Tasserは「 Zhang-server 」という名前でCASPのタンパク質構造の予測競争に勝ちました。
たとえば、インフルエンザウイルスのモデルを扱うと、表面タンパク質の1つであるノイラミニダーゼが、酵素機能(細胞膜糖タンパク質の組成におけるシアル酸の分解)を直接実行する構造の一部のみを実験的に決定するという事実に出会いました。 タンパク質のステムを形成し、ウイルスの脂質エンベロープ内のノイラミニダーゼを固定する分子の部分は、相同性によってモデル化する必要がありました。 パラインフルエンザウイルス血球凝集素-ノイラミニダーゼ( 3TSI )および膜貫通ペプチドの1つ( 2LAT )の記載されている構造をテンプレートとして使用しました。

インフルエンザウイルスのノイラミニダーゼ複合体をモデリングするためのテンプレート。 AはPDBデータベースの2AEP構造のノイラミニダーゼN2モノマーのフラグメント、Bはパラインフルエンザ血球凝集素ノイラミニダーゼ(3TSI)のステム、Cは2LAT膜貫通ペプチドです。 Dは受け取った最終モデルです。
通常、最終的なタンパク質モデルは、I-Tasserサーバーのモデルだけでなく、さまざまなテンプレートメソッドによって検出されたそのフラグメントの既知の構造を考慮して作成されます。 これを行うには、 Modellerプログラムを使用します。 1つ以上のテンプレートを使用して相同性によってモデルを構築し、追加の変更を加えることができます。たとえば、所定の場所にジスルフィド結合を作成します。
ドッキング
科学文献ではしばしば情報が不完全なウイルスの構造のもう1つの重要な側面は、個々のタンパク質間の相互作用です。 私たちの場合、個々のタンパク質のモデルのどの表面が互いに接触し、最終モデルのビリオンの他の成分に接触するかによって異なります。 相互作用に関する情報は、構造バイオインフォマティクスの明確化も可能にします。
ドッキングプログラムは、複雑な形成の自然なプロセスをモデル化せず、遅すぎてリソース集約型になりますが、最適な構造を求めて2つ以上の分子の相対位置のオプションを列挙します。 ドッキングするとき、通常、複合体内の大きな分子は受容体と呼ばれ、小さな分子はリガンドと呼ばれます。 リガンドと受容体の複合体の構造の質を評価するために、さまざまな評価関数が使用されます。 理想的には、そのような関数はシステムの自由エネルギーである必要がありますが、計算するのは複雑すぎるため、ポテンシャルエネルギー(単純に計算される)、リガンドと受容体の接触面積、研究者が分析から導き出したさまざまなルールを考慮するさまざまな経験的擬ポテンシャルが使用されます多数の複合体、および物理的な意味を持たないあらゆる種類の神秘的な用語。ただし、多数の既知の複合体でテストすると、プログラムの結果が改善されます。 現代のプログラムにおけるこのような擬ポテンシャルの最小値の探索は、通常、モンテカルロ法と遺伝的アルゴリズムのさまざまなバリエーションを使用して行われます。 現在、多くの分子ドッキングプログラムがあり(最も有名なものはDock 、 Autodock 、 GOLD 、 Flexx 、 Glide )、評価関数、最小化方法、追加機能が異なります。 同時に、検索中に、受容体分子とリガンド分子の両方が静止したままである場合があり(このタイプのドッキングはハードと呼ばれます)、コンフォメーションを多少変更します(柔軟なドッキング)。 明らかに、2番目のオプションはより多くのリソースを消費しますが、このような検索の結果は通常信じられます。 低分子をタンパク質にドッキングすることは、現在、新薬開発の標準的なステップです。 たとえば、1000万個のリガンドのドッキングを実行し、さらに実験的な作業のために最も有望な100個の化合物を選択できます。これは仮想スクリーニングと呼ばれます。
小分子の研究に加えて、ドッキングを使用してタンパク質-タンパク質およびタンパク質-ヌクレオチド複合体を構築できます。 これらの目的のために、多数のプログラムとオンラインサービスが開発されています( ZDOCK 、 pyDOCK 、 HEX )。 たとえば、 ヒトパピローマウイルス (HPV)に関する研究の過程で、L1タンパク質によって形成されるキャプシドの外層の完全な構造にもかかわらず、キャプシド内のゲノムに近い位置にあるL2タンパク質の構造に関する情報がまったくないという事実に直面しました。したがって、ペンタマーL1がL2分子と相互作用する方法に関するデータはありません。 Tasserサーバーを使用して相同性L2タンパク質モデルを構築し、HeXプログラムにドッキングしました。 ドッキング中、五量体L1は受容体として機能しました。 最適な着陸地点L2の検索が実行されたのは、その表面にありました。 同時に、すべての構造物は動かないままでした。 つまり ハードドッキング方法が使用されました。 その結果、L1とマイナータンパク質L2から組み立てられた五量体複合体のもっともらしい構造が得られました。

メインキャプシドタンパク質L1の五量体とマイナータンパク質L2との複合体(ウイルス粒子の右側のポスターに表示)。 下からの眺め(分解)と上からの眺め。 相同性とドッキングモデリング手法の組み合わせによって得られた構造。
翻訳後修飾
最後に、バイオインフォマティクスの手法は、ウイルスタンパク質の構造のどのような変化が、それらが形成される細胞自体を作るかを復元しようとすることができます。 合成後、ほとんどのタンパク質は追加の化学的翻訳後修飾(PTM)を受けますが、これはタンパク質によって実行される機能に深刻な影響を与える可能性があります。 これらの修飾には、リン酸化、ユビキチン化、グリコシル化、ニトロシル化、ギャップの導入、その他の化学変化があります。 ウイルスの表面タンパク質の多くはグリコシル化されており、この修飾はウイルスの表面タンパク質の主な機能-細胞受容体への結合-にとって直接重要です。 一方、ウイルスマトリックスのタンパク質-一部のウイルスの脂質膜の直下にある層は、膜に固定するために、たとえばミリスチン酸と関連付けられている必要があります-タンパク質と脂質の相互作用を促進する小さな疎水性分子。 したがって、私たちの研究では、タンパク質の修飾にも注意が必要です。
現在、可能なPTMの予測はかなり困難です。 既存の主な方法とサービスは、類似のタンパク質の関連する実験情報の検索、または特定のタイプの修飾に特徴的な小さな領域の研究対象タンパク質の配列の検索に基づいています。
私たちの仕事では、モデルを準備する際に、対応するUNIPROTデータベースレコードに反映された実験情報を使用します。
インフルエンザウイルス血球凝集素モデルの作業段階。 A-PDBデータベースからの3ZTJ構造の視覚化。 B-H1N1インフルエンザウイルス血球凝集素モデル。分子の膜貫通部分が完成した3ZTJとの相同性に基づいて構築されています。 C-翻訳後修飾(グリコシル化)を考慮したモデル。
分子動力学と構造最適化
最後に言及したいのは、タンパク質および特にそれらの複合体の新しいモデルを準備するとき、構造の最適化を実行する必要があるということです。 最も単純な最適化方法は、エネルギーを最小化することです。 これは、システムをローカルポテンシャルエネルギーの最小値にすばやく「下げる」ために使用されます。 この操作は、分子構造の各修正後に実行することが好ましい。 原子の重なりや不規則な結合長の出現などのトラブルを回避します。 エネルギーを最小化するさまざまな方法が、ほぼすべての分子モデリングソフトウェアパッケージで提供されています。
この方法では、予備的な非常に粗雑な最適化のみが許可されることに注意してください。 空間構造をより正確に準備するために、分子動力学法または量子力学法が使用されます。 後者は、例えば、小さなリガンド分子の構造の最適な最適化と分子間相互作用のエネルギーの最も正確な計算に使用されます。 しかし、非常に論理的な最高の精度は、より多くのリソースを消費する計算に関連しているため、大きな生体高分子に適用した場合、これらの方法は実用的ではありません。
分子動力学法により、ポリペプチドや核酸などの十分に大きな分子の構造の挙動と安定性を評価できます。
分子動力学法は、原子と分子の振る舞いおよびそれらの時間内の動きを研究することです。 分子動力学の計算により、たとえば、個々の分子とその複合体の両方の安定性を研究し、可能なコンフォメーション再編成の重要性、点突然変異の影響などを評価することができます。 分子動力学シミュレーションの結果を分析する最新の方法は、個々の原子と研究中のシステム全体の両方の時間挙動に関する最も詳細な情報を提供します。
モデルを作成したいウイルスのタンパク質をどれだけよく研究しているかに応じて、すべてのタンパク質とその相互作用のモデルを完成および最適化するためのアプローチを選択する必要があるたびに。 すべての構造が得られたら、完全なモデルの組み立てを進めることができます。 ヒトウイルスの科学的に信頼できるモデルの作成に関するシリーズの次の投稿では、これがどのように行われるかを説明します。
PS:
最後の投稿の調査でリーダーとなったトピックは、 医療解剖学的イラストです。5世紀のイラストレーターの作品における人体の研究の歴史は次の予定です。 見事な彫刻、前世紀のワックスモデル、死体の可塑剤、優れた研究者のアトラス、冷凍自殺爆撃機の階層化セクションに基づく3D再構築、インタラクティブなアプリケーション、現代の医療イラストレーターの作品。 近日公開。