理論のビット
まず、ファイルパスを取得する方法は関係ありません-少なくともQM、少なくともMM、少なくとも何らかのハイブリッド。 重要なことは、原子の正確な座標を知る必要があるということです。 軌跡であるかどうかは関係ありません-軌跡のスナップショットを1つ使用することはできますが、統計情報はこの影響を受け、結果として結果が不正確になる可能性があります。
次に、水素結合の決定に関連する質問に移ります。 水素結合の存在(または不在)をいくつかの可能な方法(およびそれらの組み合わせ)で決定することが提案されています。例えば:
- 異なる分子の酸素原子と別の分子の水素原子と酸素原子の間の距離は、基準値よりも小さくする必要があります
- 異なる分子の酸素原子間の距離とHOO角度は、基準値よりも小さくする必要があります
- エネルギー基準
何を考慮する必要がありますか?
ご存知のように、最新のリソースでは、かさばるシステム(1モルの分子を含む単位セル)をシミュレートできないため、 周期的な境界条件を使用する必要があります 。 実際の無限システムは、すべてが同じセルに囲まれた空間セルに配置された同一の有限システムのセットに置き換えられ、1つのセル内の分子は隣接するセル内の分子と相互作用します。 実際には、このような分析では、セルをすべての方向に変換する必要はまったくなく、推定スペースが27倍になります(1つのセルではなく3 ^ 3 = 27になります)。 各方向にrib骨の半分をブロードキャストするように制限できます。 この場合、推定スペースを8倍だけ増やすことを示すのは簡単です。 同時に、明示的に翻訳し、新しい座標を変数にスコアリングするか、単に分析で翻訳の可能性を「覚えておく」ことができます。 私のプログラムは最初のオプションを実装しています。 ちなみに、この場合、 翻訳された細胞内の分子との接続は、元の細胞内の分子に移さなければならないことに留意してください 。
2つ目の問題は、2つの官能基を持つ分子がある場合はどうなるかということです。 たとえば、水素結合の場合-エタンジオール。 またはさらに楽しい-水。 分子の数という追加のラベルを導入することにより、この機会を考慮することをお勧めします。 つまり、個別のグループではなく、すぐに分子を操作します。
3番目の問題は、OH基を持つシステムだけでなく、たとえばクロロベンゼンでも同様の凝集が観察されることです。 私は、そのような関連がコロイド系におけるミセル形成に似ていることだけに注意し、その方法と理由については詳しく述べません。 この場合、塩素と塩素の距離を分析して、分子が結合するかどうかを判断できます。
実装
すぐにコメントをいくつかします。 私からのプログラマーは、完全に曲がっているだけではありません。確かに、何かをもっとうまくできるからです。 最初はブロック図を作成することを考えましたが、面倒すぎることがわかりました。
ソースデータ:
- メタノール、酸素原子、およびそれに関連する水素の例では、私たちにとって関心のある原子の座標(翻訳されたものを含む)
- 特定の官能基が属する分子を示すラベルの配列。
- 2つのパラメーター-参照分子間距離OHおよびOO
結合分子を検索するためのアルゴリズム:
- すべての分子(翻訳されたものを含む)を反復する2つのネストされたループ-2番目のループは、当然、次の分子から始まります(同じことを2回考慮する必要があるのはなぜですか?)。
- 酸素原子間の距離を考慮し、分子内のすべてのグループについて、それらの間で最小値を見つけます。 リファレンスで確認します。 少ない場合は、次に進みます。
- 前のステップの実装。ただし、すでに酸素および水素原子用。 つまり、すべての可能なもの(1つの酸素、たとえば水中の2つの水素の場合)を最初の分子の酸素原子(前のステップで定義)と2番目の分子の水素(前のステップで決定された酸素に関連付けられている)の間の距離と見なし、最小。 リファレンスで確認します。 少ない場合は、次に進みます。 それ以上の場合は、最初に反対のオプション-最初の分子の水素原子と2番目の分子の酸素の間の距離-をチェックします。 少ない場合は、さらに進んでください。次の分子に進みます。
- これらの分子は接続されていると書きます(必要に応じて結合の長さも)、次の手順に進みます。 この場合、翻訳された分子とのすべての通信は、元のセルの分子に記録されます。
出力アルゴリズム:
- Trueで元のセルに各分子のバイナリラベル(つまり、翻訳されたものなし)を配置します。 元のセル内の分子に対してループが繰り返されました。
- Trueというラベルの付いた最初の分子を取得し、Falseというラベルを付けます。 それを凝集体の配列に書き込み、次にラベルがTrueであり、それに関連付けられているすべての分子を書き留めて、それらをFalseと見なします。 新しい接続がなくなるまで
、青色に変わるまで続けます。 - すべての分子にFalseラベルが付くまで、前の段落を繰り返します。
次に、便利な形式で凝集体の配列を書き出します。 最後に、一般的な統計(軌跡のすべてのポイントで平均化)を持つブロックを追加します。 たとえば、酸素原子間の結合の統計を追加できます。
他に何を追加しますか?
1つ目は、従業員のサイクルの有無に関する相互分析です。 証拠に触れることなく、私はそれに注意します。 1つの分子に関連付けられた分子に0、2つの-1 -1などの重みを割り当て、グラフの合計重量を計算(関連付け)すると、グラフのサイクル数を決定できます。
- グラフの重みPがN-2(Nはグラフの頂点の数(凝集体の分子))の場合、グラフにはサイクルがなく、分岐構造または線形構造のいずれかです。
- グラフの重みPがN-2より大きい場合、グラフには(PN)/ 2 + 1の量のサイクルがあります。
2番目ははるかに楽しいです。 こことここにオリジナルの記事があります 。 ポイントは、2つのグラフがある場合、それらが同型であるかどうか(同じ±順列の意味で)を比較し、一方のグラフから他方のグラフが取得される順列を決定できることです。 ただし、メモリが適切に機能する場合、これらの記事ではアルゴリズム自体にタイプミスがありました。注意してください。
私たちのシステムに適用されると、基本的なグラフのデータベースを作成し、特定のグラフに準拠しているかどうかを結果として関連付けを確認できます。 統計を実施することもできます。
残念ながら、このブロックは実装していません。 2つのグラフの同型をチェックするプログラムのみが利用可能です。
さて、3番目。 1つの分子の分析は、軌跡全体にわたるさまざまな凝集体におけるその存在のダイナミクスです。
さらに
リンクからプログラムのソースコードを取得できます。 すべてがCで実装されています。残念ながら、コードにはコメントがありません。
STAT_GENプログラムは、TINKERの軌跡ファイルに似たファイル形式で動作しますが、1つの例外があります。さまざまな原子の相互接続に対応するブロックはなく、分子の数を示す列が追加されています。
AGLプログラムは、パスファイルから個々の凝集体を引き出し、PDB形式に書き込みます。
グラフプログラムは、グラフの同型を分析します。
残念ながら、LInuxに移行した後のWindowsでのこれらのプログラムの操作性(たとえば、conio.hライブラリがあるため、もともとWindowsで書かれていました)は、それが何であるかわからないためテストされていません。
出力では、次の図のようなものを取得できます(私のプレゼンテーションの1つから)。
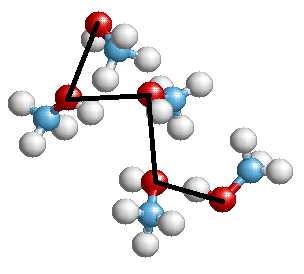
または、グラフ形式の別の構造:
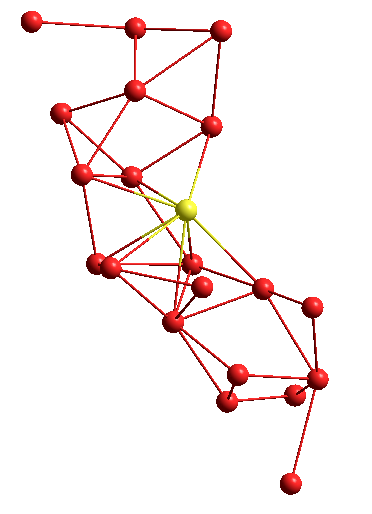
ご清聴ありがとうございました!