 最も難しいものについて、できるだけ早く話そうとする努力の中で、明らかに最も単純なものを忘れます。 そして、私の場合、単純なだけでなく、重要なリンクについても。 因果関係はわずかに壊れています。 私の以前の記事( 1、2、3、4 )では、数学的な側面とプログラミングについて説明していますが、実際には生物学はありません。 したがって、この記事では、分子生物学のどの部分で、私が説明するプログラムとアルゴリズムを発見、予測、確認、解決しようとしているのかについて説明します。
最も難しいものについて、できるだけ早く話そうとする努力の中で、明らかに最も単純なものを忘れます。 そして、私の場合、単純なだけでなく、重要なリンクについても。 因果関係はわずかに壊れています。 私の以前の記事( 1、2、3、4 )では、数学的な側面とプログラミングについて説明していますが、実際には生物学はありません。 したがって、この記事では、分子生物学のどの部分で、私が説明するプログラムとアルゴリズムを発見、予測、確認、解決しようとしているのかについて説明します。
表面に卵子と精子の画像が描かれた絵は、私が見逃したステージを象徴しています。 興味深い事実は、2つの細胞の結合により、人体に約10兆個の細胞が生じることです。
分子生物学への短いエクスカーションから始めましょう。 複雑なことは簡単な言葉で説明し、詳細は省略します。 DNAは細胞内にあります。 私たちはDNAとその上で行われるプロセスを正確に検討していますが、細胞内の正確な位置にはあまり関心がありません。 使用する真核細胞の例が与えられることを明確にする必要がありますが、 原核生物でも多くのことがうまくいく可能性が高いです。
細胞内のすべてのプロセスはDNAから始まり、 ヌクレオチド配列が存在するのはDNA上であり、将来、そのコピーが細胞内の反応と形質転換の原因となります。 転写と呼ばれるコピープロセスに興味があります。 転写産物と得られる最終産物の量をコピーするプロセスは、 遺伝子発現と呼ばれます。 しかし、遺伝子発現と呼ばれ、転写産物のコピー数を正確に測定します(得られた最終産物の数に関係なく)。
以下は、「細胞内で発生する4つの基本的な遺伝プロセス」と呼ばれる生物学の教科書の図です(図1を参照)。 現在、この図に示されている数字1と2の間のプロセスに取り組んでいます。学習に役立つ技術はDNA-seqおよびRNA-seqと呼ばれます。 これらのテクノロジーは、個別に使用することも、一緒に使用することもできます。 この記事では、DNAシーケンスのメカニズムについて詳しく説明します。
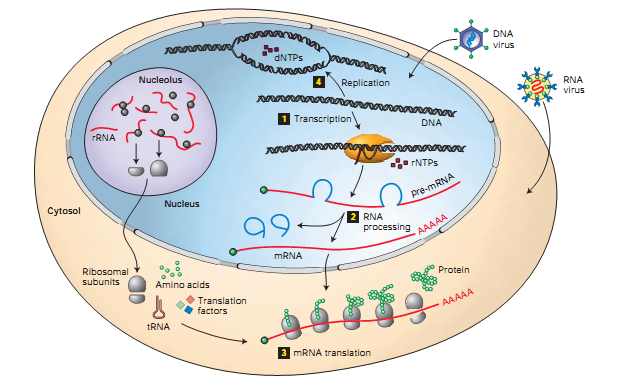
図 1
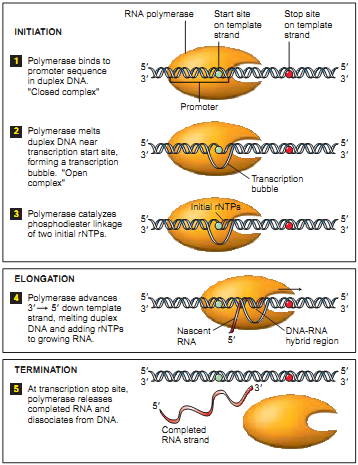 DNA-seqの特殊なケースとして、「DNA-seq Pol II」を検討してください(「 ZINBAを使用する準備をする 」も参照)。 その助けを借りて得られた結果の理解は、転写のプロセスに基づいています。 転写の過程で重要な役割を果たしているのは、RNAポリメラーゼタンパク質( RNA Polymerase )です。 ポリメラーゼは、プロモーターと呼ばれる特別なDNAサイトに固定されています(図2.1を参照)。 転写プロセスの準備操作が行われている間(開始段階)、ポリメラーゼはプロモーターに付着したままです。 ポリメラーゼは、サイトの開始点からコピーされた上部ヘリックスのコピーを作成し始めます(コピーされたチェーンの開始点、図の青い点で示されています)。 次に、毎分約1000塩基の速度(37°Cの温度)で移動し、DNAの一部を停止部位にコピーします(コピーされたチェーンの終わりは図の赤い点で示されています)。 プロモーターの位置では、ポリメラーゼがほとんどの時間に位置し、コピーされたDNAの各部位にはより少ないことを再度強調します。 さらなる研究のために、科学者はプロモーターと開始部位に興味があります。これらは転写の開始に必要な条件です。 プロモーターには、コピーの数に関する情報も含まれており、これも研究の重要な目的です。
DNA-seqの特殊なケースとして、「DNA-seq Pol II」を検討してください(「 ZINBAを使用する準備をする 」も参照)。 その助けを借りて得られた結果の理解は、転写のプロセスに基づいています。 転写の過程で重要な役割を果たしているのは、RNAポリメラーゼタンパク質( RNA Polymerase )です。 ポリメラーゼは、プロモーターと呼ばれる特別なDNAサイトに固定されています(図2.1を参照)。 転写プロセスの準備操作が行われている間(開始段階)、ポリメラーゼはプロモーターに付着したままです。 ポリメラーゼは、サイトの開始点からコピーされた上部ヘリックスのコピーを作成し始めます(コピーされたチェーンの開始点、図の青い点で示されています)。 次に、毎分約1000塩基の速度(37°Cの温度)で移動し、DNAの一部を停止部位にコピーします(コピーされたチェーンの終わりは図の赤い点で示されています)。 プロモーターの位置では、ポリメラーゼがほとんどの時間に位置し、コピーされたDNAの各部位にはより少ないことを再度強調します。 さらなる研究のために、科学者はプロモーターと開始部位に興味があります。これらは転写の開始に必要な条件です。 プロモーターには、コピーの数に関する情報も含まれており、これも研究の重要な目的です。
沈着を伴うDNA-seq Pol IIプロセスは、いくつかの段階で行われます。1.ポリメラーゼの現在の位置を化学的または熱的に固定します(ポリメラーゼを固定すると、プロセスが停止し、DNAに沿ってポリメラーゼが移動しなくなります); 2.任意の方法でDNAを切断します(超音波、 MNaseなど)(ほとんどの場合、ポリメラーゼで保護されていないDNAセクションを切断します)。 3.クロマチン免疫沈降(Pol IIを除く他のすべてのタンパク質に対して免疫性のある抗体)を使用して、ポリメラーゼを含む領域を選択します。 その結果、約150塩基の長さのフラグメントを取得し、それをシーケンサーに送信します。 ポリメラーゼ切断は厳密にはエッジで行われないことを強調する必要があります。 したがって、プロセスは次のようになります。数百万の同じDNAを異なる細胞から調製し、固定ポリメラーゼはそのほとんどがプロモーター上にあり、一部は遺伝子上にランダムに分布しています。 次の図で説明するプロセスのデジタル化の結果は、記事の1つで示されています(図3を参照)。 提案されたプロモーター(高密度読み取り)を定義するリードは緑色の丸で囲まれ、その後ろには動作中のポリメラーゼに対応する低密度読み取りがあります。
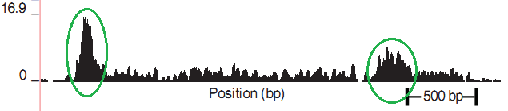
図 3
次に、他のいくつかのタンパク質について簡単に説明します。 図4の上部では、DNAがどのようにパックされているかを確認できます(図4.1を参照)。また、図の下部では、上記の転写プロセスの詳細を説明しています(図4.2を参照)。 図4.1に見られるように、DNAパッケージは非常に高密度で、ヌクレオソームの周りに二重らせんが巻かれており、DNA鎖の長さは約146塩基です。 ヌクレオソームはタンパク質-ヒストンで構成されています。 ヒストンにはさまざまな修正を加えることができますが、ヒストンの修正は別の大きな記事の主題です。 抗体は、ヒストンの特定の修飾用に開発されており、沈殿した(ろ過された)ヒストンのこの修飾の周りを包むのは、まさにDNAのセクションです。 変更表記の例を次に示します:H3K4Me3。 ヒストンH3の4番目の位置にあるリジンのトリメチル化として読み取られます。 このようなヒストンの修飾は多く存在する可能性があり、それらはすべてDNAによって散在しています。そのため、H2K27Me3(ヒストン3のリジン27のトリメチル化)を示す図2の記事「 ZINBAでの作業の準備 」では、このような頻繁なピークヒープが観察されています。
DNA-seqテクノロジーは沈殿なしで存在します。たとえば、DNase酵素を使用してDNAを単純に切断すると、膨大な数のフラグメントが得られます。 ほとんどの場合、DNaseはヌクレオソーム間を切断するため、断片はヌクレオソームを包むターンに対応します。 結果のフラグメントはシーケンサーに送信されます。 ディープシーケンス(膨大な数の断片、約1億の読み取りがシーケンサーに送られますが、それは高価な喜びです)を使用すると、結果のランドスケープからタンパク質がどこにあるかを認識できると考えられています。 さらに、風景は火山のように見え、上部の小さな窪みはリスに対応するはずです。 ほとんどの場合、DNaseメソッドは、この酵素に敏感なDNA領域を見つけるために使用されます。 最高のカットされている領域。
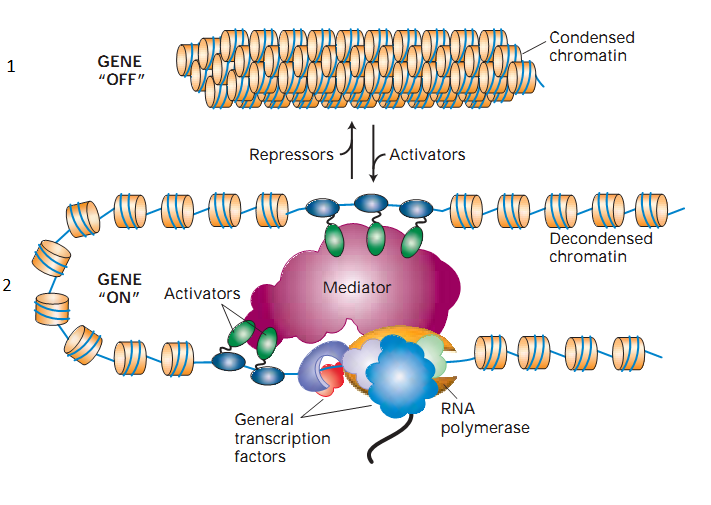
図 4
図の下部には、RNAポリメラーゼと、その中およびDNA上に配置されたマルチカラーの雲と楕円があります。 これらの追加要素は転写因子と呼ばれ、遺伝子発現の調節に重要な役割を果たします。 転写因子は、他の転写因子の追加の可能性を増加、減少、または単純にブロックすることができ、それにより暗黙的に発現の調節につながります。 それらはタンパク質でもあります。 他のすべてのタンパク質と同様に、抗体はそれらに対して開発されます。 CTCFタンパク質を転写因子の例として見てみましょう。その役割の1つは、他の転写因子の働きをブロックすることです。 それを用いたDNA-seq実験については、記事「 ZINBAを使用する準備 」で説明されています。対応するCTCFクロマチン免疫沈降の状況を次の図に示します。 ご覧のとおり、このタンパク質によって保護されている領域は小さいため、周辺の広がりは大きくなく、150-200bpのみです。
DNA-seq実験の種類のスキーム例(配列決定法):
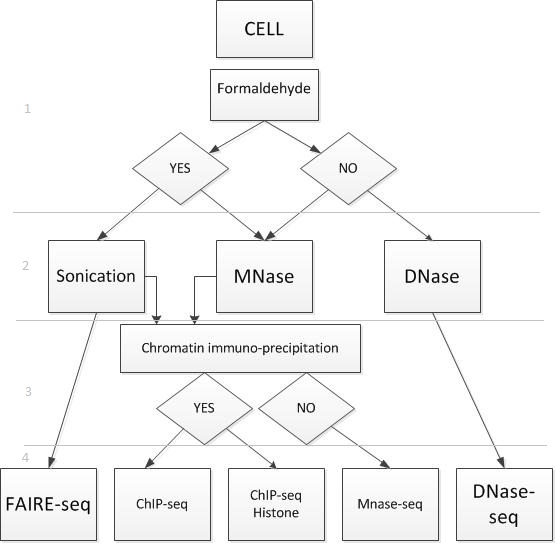
図 5
スキームを4つの条件付きレベルに分割しました。 最初のレベルは固定で、ホルムアルデヒドの有無にかかわらず実行できます。 2番目のレベルは、DNAを切断する方法です。 3番目のレベルはサイズによるろ過です。これに加えて、クロマチン免疫沈降を使用してろ過を実行できます。 4番目のレベルはシーケンス方法です。
したがって、実施された各タイプの実験は、シーケンス方法に対応しています。 シーケンスの方法に応じて、さまざまなタイプのフラグメントが作成されます。 結果がタンパク質を含む断片である実験を考えてください。 タンパク質は、ピースの中央またはピースの両側のいずれかに配置できます。 シーケンサーは、セグメント全体をデコードするのではなく、各フラグメントのスパイラルの端の5 '側からセグメントのほんの一部をデコードします。 デコードされた断片は読み取りと呼ばれます。 図6では、各スパイラルの5 '端に赤い点が付いています。 青と赤の矢印は、読み取りの開始を示します。 これらの始まりは、各ヘリックスの5 '端からタンパク質の境界までランダムに分布しています。
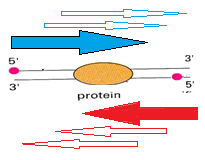
図 6
多くの初期分子が実験に参加し、それらのDNAは特定のタンパク質近傍で切断され、多くの読み取りが(タンパク質の1つの)座標軸に表示されると、次のような画像が得られます。

図 7
座標軸の上(濃い青)には、上部スパイラル(「+」ストランド)からの読み取り値が表示され、軸(水色)の下には、下部スパイラル(「-」ストランド)からの読み取り値が表示されます。 タンパク質が不可能だった場所を正確に言うと、タンパク質結合中心はこれらの2つのピークの間にあると想定されます。
結果の画像は信号に似ているため、信号を分析するためのいくつかのアルゴリズムがデータに適用され始めました。そのうちの1つは、この記事habrahabr.ru/blogs/algorithm/135281- 「隠れマルコフモデルとは」に記載されています。
この記事の後、主題がより明確になり、さらなる研究がわずかな恐怖を引き起こさないことを願っています。 誰かが資料のより詳細なプレゼンテーションに興味を持っている場合、分子生物学の基礎が非常に有益でアクセス可能な、ロディッシュ編集の素晴らしい分子細胞生物学の本があります。いくつかの図はそれから借用されています。
レビューは、オハイオ州シンシナティのアンドレイ・カルタショフによって作成されました。porter@ porter.st。